Table 39-2
2004 WHO Classification Of RCC

Approximately 2% to 5% of kidney cancers are linked to a recognized cancer predisposition syndrome, and there is at least one such syndrome for each of the major histological kidney cancer subtypes (see Table 39-1). Germline VHL mutations cause VHL disease, in which ccRCC is a dominant feature, whereas germline c-Met and fumarate hydratase mutations are linked to hereditary Type 1 and Type 2 pRCC, respectively. Germline FLCN mutations cause familial oncocytomas together with an increased risk of renal cell carcinoma (especially chRCC).
von Hippel-Lindau Disease
von Hippel-Lindau disease is an autosomal dominant disorder caused by germline loss-of-function mutations of the von Hippel-Lindau tumor suppressor gene (VHL), which is located on chromosome 3p25, and affects about 1 in 35,000 people. 7 Most VHL patients, however, will exhibit some manifestations of their disease by early adulthood. Hemangioblastomas and kidney cancer are the two leading causes of death in this population. All patients with a clinical diagnosis of VHL disease harbor a VHL mutation. A number of genotype-phenotype correlations have been described in this setting, however, with respect to the risk of developing specific tumors such as kidney cancer and pheochromocytoma. Type 1 VHL disease is associated with low risk of pheochromocytoma and Type 2 with a high risk of pheochromocytoma. Type 2 disease is subdivided into Type 2A (low risk of kidney cancer), Type 2B (high risk of kidney cancer), and Type 2C (pheochromocytoma only).
Somatic VHL Mutations and Hypermethylation in Sporadic Clear Cell Renal Carcinoma
Of sporadic ccRCCs, 40% to 80% have inactivated both VHL alleles (maternal and paternal), most often as a result of a somatic intragenic mutation affecting one VHL allele and a large chromosome 3p somatic deletion spanning the other (Table 39-3 ). In short, the VHL tumor suppressor gene, like the RB1 tumor suppressor gene, conforms to the Knudson two-hit model of carcinogenesis. An additional 10% to 20% of clear cell renal carcinomas have not sustained VHL mutations but fail to produce normal levels of the VHL mRNA (and hence protein) because the maternal and paternal VHL loci are hypermethylated. Some other clear cell carcinomas display a transcriptional signature suggestive of VHL inactivation despite an apparent absence of VHL mutations or hypermethylation.
Table 39-3
Somatic Genetic Alterations in Sporadic Renal Carcinomas
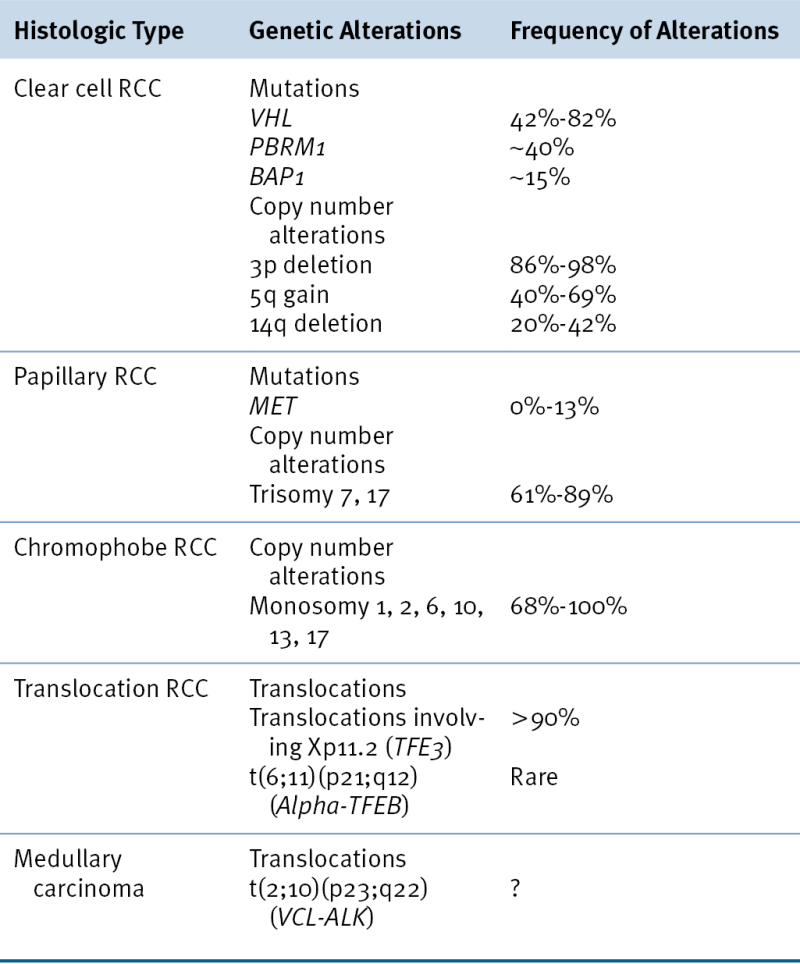
VHL Protein Function
The VHL gene encodes two proteins by virtue of two alternative, in-frame, translation initiation codons. 8–10 Both protein isoforms behave similarly in most (but not all) assays performed to date, and the VHL mutations linked to kidney cancer almost invariably affect both isoforms. pVHL dynamically shuttles between the cytoplasm and the nucleus, although at steady state most of the protein is cytoplasmic. pVHL is a multifunctional protein whose function relates to its role as the substrate recognition module of an E3 ubiquitin ligase complex that also contains elongin B, elongin C, Cul2, and Rbx1 11 (Figure 39-1 ). The crystal structure of pVHL bound to elongin B and elongin C reveals that pVHL has two subdomains, called the alpha domain and the beta domain, which are hotspots for mutations in VHL disease and which are, directly or indirectly, affected by kidney cancer–associated mutations. A number of putative pVHL substrates have been identified, including members of the HIF transcription factor family.
The HIF Transcription Factor
HIF (hypoxia-inducible factor) is a heterodimeric transcription factor consisting of an alpha subunit and a beta subunit, both of which are members of the basic Helix-Loop-Helix-PAS domain family. 12,13 There are three human HIFα (HIF1α, HIF2α, HIF3α) and three human HIFβ (often referred to as ARNTs for aryl hydrocarbon receptor nuclear translocators) family members.
In the presence of oxygen, HIFα is hydroxylated on one (or both) of two prolyl sites by members of the EglN (also called PHD) prolyl hydroxylase family 14 (see Figure 39-1). Hydroxylation of either of these sites generates a binding site for pVHL, which then directs the polyubiquitylation of the HIFα subunit. Once ubiquitylated, HIFα is then degraded by the proteasome. Under low-oxygen conditions EglN activity is impaired because these enzymes require oxygen, in addition to 2-oxoglutarate and reduced iron, in order to function. This leads to stabilization and accumulation of HIFα, which then binds to HIFβ, translocates to the nucleus, and activates 100 to 200 genes that contain hypoxia-response elements and promote adaptation to hypoxia. In contrast to HIF1α and HIF2α, most of the HIF3α isoforms detected in cells, which arise because of alternative splicing of the HIF3α mRNA, lack transactivation capability and may, in fact, serve to blunt HIF1α and HIF2α activity.
HIF and Kidney Cancer
In pVHL-defective ccRCC, polyubiquitylation of HIFα is impaired, leading to inappropriate activation of HIF target genes under normoxic conditions. HIF activates a number of genes that are believed to play, or suspected of playing, roles in kidney carcinogenesis, including TGFα, Cyclin D1, SDF1, CXCR4, MMP family members, PDGF B, and VEGF. The increased expression of factors such as VEGF likely contributes to the highly angiogenic nature of clear cell renal carcinomas and their responsiveness to VEGF blockade. The overproduction of the canonical HIF target erythropoietin accounts for the association of VHL-associated neoplasms, including renal carcinomas, with paraneoplastic erythrocytosis. 15 HIFα, acting without its partner HIFβ, can also activate Notch signaling, which has been implicated in renal carcinogenesis.
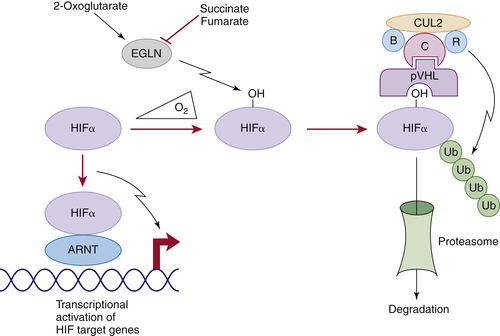
Figure 39-1 Regulation of HIF by pVHL In the presence of oxygen, HIFα is hydroxylated on one (or both) of two conserved prolyl residues by EglN family members. This creates a binding site for pVHL, which is stably bound to elongin B (B), elongin C (C), Cul2, and Rbx1 (R). This complex then polyubiquitylates HIFα, marking it for proteasomal degradation. Under low-oxygen conditions, or in cells lacking pVHL, HIFα binds to ARNT, translocates to the nucleus, and activates HIF target genes such as VEGF. EglN family members require 2-oxoglutarate, in addition to oxygen, and are inhibited by succinate and fumarate.
Several lines of evidence suggest that deregulation of HIF, and particularly deregulation of HIF2α, plays a causal role in pVHL-defective renal carcinoma. To date virtually all kidney cancer–associated VHL mutations, whether germline or somatic, compromise pVHL’s ability to regulate HIF, and the risk of developing kidney cancer in VHL families correlates well with the degree to which their mutant VHL alleles deregulate HIF. Close examination of preneoplastic renal lesions in VHL patients reveals that the appearance of HIF2α is associated with histological changes indicative of incipient malignant transformation.
In stark contrast to HIF2α, HIF1α appears to act as a renal cancer suppressor. HIF1α resides on chromosome 14q, which is frequently deleted in kidney cancers. HIF1α mRNA and protein is decreased in a sizable fraction of clear cell renal tumors, and HIF1α-responsive mRNAs are diminished in 14q-deleted renal tumors. It should be noted, however, that most 14q-deleted clear cell renal tumors, in contrast to cell lines, appear to retain a wild-type HIF1α allele. It is therefore possible that haploinsufficiency of HIF1α promotes tumor growth in vivo and that reduction to nullizygosity occurs during tumor progression (most cancer lines are derived from metastatic lesions) or establishment and propagation of cell lines. Although rare, intragenic HIF1α mutations have been reported in kidney cancers. Collectively, these findings suggest that HIF1α is a target of the 14q deletions that are typical of clear cell renal carcinomas.
The differential effects of HIF1α and HIF2α in pVHL-defective tumor growth likely reflect both qualitative and quantitative differences. For example, the genes regulated by HIF1α and HIF2α overlap but are clearly not identical. Perhaps HIF1α preferentially induces the expression of a renal tumor suppressor, or suppressors, whereas HIF2α preferentially induces the expression of one or more downstream oncogenes. Moreover, HIF1α can potentially antagonize HIF2α with respect to the induction of shared targets under normoxic conditions in pVHL-defective cells because transcriptional activation by HIF1α, in contrast to HIF2α, is enfeebled by the oxygen-dependent asparaginyl hydroxylase FIH1.
HIF-Independent pVHL Functions
pVHL has a number of other functions that appear to be at least partially HIF-independent and that might contribute to renal cancer suppression. For example, pVHL promotes fibronectin matrix assembly, maintains microtubule stability and a specialized structure called the primary cilium, suppresses the activating phosphorylation of the NFκB agonist CARD9, suppresses signaling by HGF and c-MET, suppresses Wnt signaling, inhibits atypical PKC activity, promotes the Cbl-independent ubiquitylation of EGFR, and regulates senescence. The ciliary defect in pVHL-defective renal epithelial cells is particularly intriguing because renal cysts are a prominent feature of both VHL disease as well as the other primary ciliopathies.
Mouse Models of VHL Disease
There are currently no genetically engineered mouse models that develop VHL–/– clear cell renal carcinomas. VHL–/– embryos die before term, and VHL+/– do not develop the usual stigmata of VHL disease. They do, however, develop hepatic blood vessel lesions that are associated with stochastic loss of the remaining wild-type VHL allele and HIF activation. VHL has also been inactivated using conditional alleles in a variety of mouse tissues, leading to phenotypes such as renal cysts (kidney), hepatosteatosis and hemangiomas (liver), cardiomyopathy (heart), and increased angiogenesis and weight loss (skin). Concurrent inactivation of PTEN in the kidney exacerbates the renal cyst phenotype but still does not lead to renal tumor formation. In most of the models tested so far, deregulation of HIF2α appears to be necessary and sufficient for the pathology observed on after pVHL loss in mouse tissues.
Other Hereditary Forms of Cancer
Individuals with germline activating c-Met mutations are highly predisposed to Type 1 papillary renal carcinoma, often associated with duplication of the mutant c-Met allele located at 7q31 16,17 (see Table 39-1). c-Met encodes a receptor tyrosine kinase that influences cellular proliferation, survival, invasion, and metastasis. Somatic c-Met mutations are relatively rare in sporadic papillary renal carcinomas (see Table 39-3).
Germline loss-of-function fumarate hydratase (1q42) mutations cause a syndrome characterized by Type 2 papillary renal carcinoma, cutaneous leiomyomata, and uterine fibroids, whereas succinate dehydrogenase subunit mutations cause familial paragangliomas and, rarely, clear cell renal carcinomas (see Table 39-1). Inactivation of fumarate hydratase and succinate dehydrogenase leads to the intracellular accumulation of fumarate and succinate, respectively, which competitively inhibit 2-oxoglutarate-dependent enzymes including the EglN prolyl hydroxylases (see Figure 39-1). Accordingly, HIFα levels are increased in FH–/– and SDH–/– tumors. It has also been shown that the secondary accumulation of succinate in fumarate hydratase–deficient cells can also, through covalent linkage to a cysteine residue, inactivate the KEAP tumor suppressor protein, leading to deregulation of the NRF2 transcription factor.
Inactivating germline mutations of FLCN (17p11.2) cause Birt-Hogg-Dube syndrome, which is characterized by a variety of dermatological lesions, including fibrofolliculomas; renal tumors including oncocytomas and chromophobe tumors and, less commonly, papillary and clear cell carcinomas; and pulmonary cysts (see Table 39-1). The FLCN gene product, Folliculin, binds to FNIP1 and FNIP2 and is believed to modulate nutrient sensing by the mTOR signaling pathway. Folliculin also appears to suppress TGFβ as well as the expression of TFE3, the common partner in kidney cancers linked to Xp11.2 translocations. Inactivating mutations of either TSC1 (9q34), which encodes hamartin, or TSC2 (16p13), which encodes tuberin, cause tuberous sclerosis. Tuberous sclerosis is associated with cutaneous, neurological, and renal abnormalities. Renal abnormalities include angiomyolipomata and, less commonly, clear cell renal carcinoma (see Table 39-1). Hamartin and tuberin form a complex that inhibits the mTOR kinase and regulates VEGF via both mTOR-dependent and independent pathways (Figure 39-2 ). Somatic TSC1 mutations have been described in sporadic ccRCC, as have mutations in other genes whose products link growth factor signaling, via hamartin and tuberin, to mTOR activity (see later discussion). Inactivation of the hamartin/tuberin complex in mice causes renal cysts, and a germline TSC2 mutation in the rat (Eker Rat) causes clear cell carcinoma.
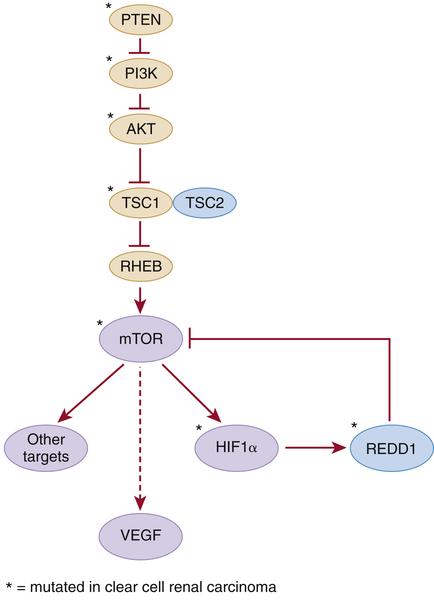
Figure 39-2 Kidney cancer mutations with an impact on mTOR Proteins that have been targets of mutations in kidney cancer are indicated by asterisks.
Some families predisposed to clear cell carcinoma carry germline translocations involving chromosome 3 and variable partners (see Table 39-1). Loss of the derivative chromosome 3p segment is believed to cause loss of tumor suppressors, such as VHL, located between 3p21 and 3p25.
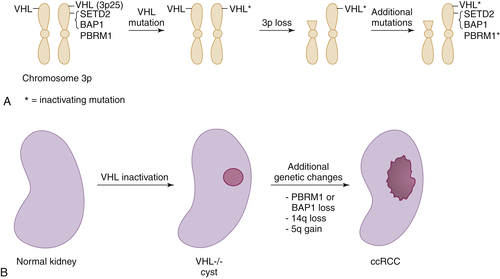
Figure 39-3 Multistep kidney carcinogenesis (A) Multiple renal cancer suppressors are located on chromosome 3p, including VHL at 3p25 and SETD2, BAP1, and PBRM1 on 3p21. For this reason three genetic “hits” can inactivate two tumor suppressors (and four “hits,” three tumor suppressors) provided one of the hits is chromosome 3p loss. In the example shown both VHL and PBRM1 have been inactivated. (B) pVHL loss leads to preneoplastic renal lesions, such as VHL–/– renal cysts. In VHL disease the kidney is germline VHL+/–, whereas in sporadic kidney cancers the kidney is germline VHL+/+. Additional genetic changes, such as PBRM1 or BAP1 mutations, loss of chromosome 14q, and gain of chromosome 5q, are linked to progression to clear cell renal carcinoma.
Common Somatic Alterations in Clear Cell Renal Carcinoma
Copy Number Changes
The most common copy number changes in clear cell renal carcinoma relate to loss of chromosome 3p, which is observed in more than 90% of these tumors (see Table 39-3). Chromosome 3p harbors several tumor suppressor genes, including the VHL gene at 3p25, and PBRM1, BAP1, and SETD2 at 3p21 (Figure 39-3 , A). Other common copy number changes in clear cell renal carcinoma include amplification of chromosome 5q and 7q and loss of chromosome 14q and 6q. Loss of chromosome 3p, gain of 5q, and loss of 14q are seen more commonly in clear cell renal carcinoma than in other cancers.
Inactivation of Tumor Suppressor Genes
PBRM1
PBRM1, located on 3p21 (see Figure 39-3, A), is mutationally inactivated in about 40% of ccRCC (see Table 39-3), typically as a result of truncating mutations of one allele coupled with loss of the remaining allele as a consequence of chromosome 3p loss. PBRM1 mutations are not mutually exclusive with mutations of the neighboring gene SETD2, indicating that the two genes are not wholly redundant. PBRM1 encodes a large protein called BAF180, which contains six bromodomains, 2 bromo-adjacent homology domains, and an HMG DNA-binding domain. BAF180 is a component of the PBAF SWI/SNF nucleosome remodeling complex and is therefore likely to affect transcription by altering chromatin structure so as to alter its accessibility to various transcription factors (Figure 39-4 , A).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


