Fig. 1
Pre-receptor regulation of glucocorticoid availability governs access to bind and activate the glucocorticoid receptor
Circulating GC levels are tightly controlled by the hypothalamo–pituitary–adrenal (HPA) axis , which regulates secretion from the adrenal glands via a classical negative feedback loop. Healthy adults secrete 10–15 mg cortisol/day [2] and the majority is bound to cortisol-binding globulin (CBG). Estimates suggest that only 5 % of circulating cortisol is “free” and biologically active [3, 4]. The half-life of free cortisol is brief (only a few minutes) whereas protein-bound cortisol has a much longer half-life between 70 and 120 min [4–6]. Importantly the biological availability of GCs represents a balance between synthesis/secretion and metabolism/clearance.
Within GC target tissues, there is an added layer of complexity to the regulation of GC action. Cortisol delivered from the circulation into cells can be subjected to a series of metabolic pathways which are able to modify the access of the active ligand, cortisol, to the GR, the so-called prereceptor regulation (Fig. 1).
Once inside the cell, a series of enzymes are able to metabolize cortisol and these include the 11β-hydroxysteroid dehydrogenases (11β-HSD1 and 2) and the A-ring reductases (5α-reductase type 1 [5αR1] and 2 [5αR2] and 5β-reductase). All have tissue-specific patterns of expression and all have been implicated in the pathogenesis of various conditions (Fig. 2). Within this chapter we will describe the enzymes involved and summarize on a tissue-by-tissue basis the contribution of each enzyme system to the regulation of GC actions.
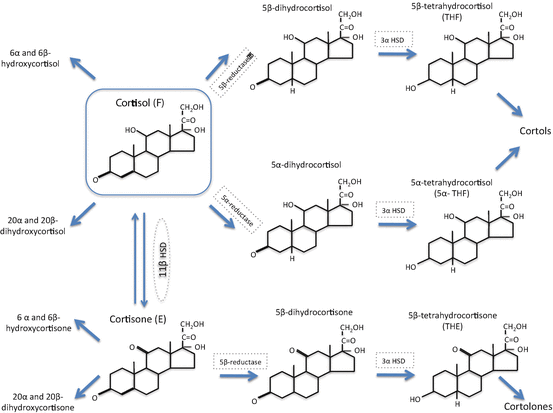
Fig. 2
The metabolism of cortisol
11β-Hydroxysteroid Dehydrogenase
11β-Hydroxysteroid Dehydrogenase Type 1
GCs were identified more than 60 years ago and were heralded as a potentially curative treatment for many diseases [7]. Kendall et al. published the discovery of what they believed to be a treatment that could reverse rheumatoid arthritis in the 1950s [8]. They identified Compound E, now recognized to be cortisone, an inactive GC metabolite that requires reactivation to cortisol (Compound F), to allow it to bind and activate the GR. It is now recognized that the enzyme responsible for the conversion of cortisone to cortisol is 11β-HSD1. The hydroxyl group at C11 is crucially important for cortisol to be active [9, 10]. 11β-HSD1 is a member of the short-chain dehydrogenase/reductase (SDR) superfamily of enzymes which are NADP(H)-dependent enzymes, which have a fundamental role in the regulation of hormone signaling with in excess of 3000 family members [11].
11β-HSD1 was purified and cloned from rodent tissues in the 1980s [12, 13]. In humans the gene that encodes the protein, HSD11B1 , is located on chromosome 1, is 30 kb in length, and has 6 exons and 5 introns. 11β-HSD1 comprises 292 amino acids and shares 77 % homology with rat amino acid sequence [14]. Human 11β-HSD1 was cloned in 2002, exists as a dimer, and is bound to the endoplasmic reticulum (ER) with its catalytic domain within the ER lumen [15–17].
11β-HSD1 is a bidirectional enzyme , which in vivo acts primarily as an oxoreductase, converting inactive cortisone (11-dehydrocorticosterone in rodents) to active cortisol. The catalytic directionality of the enzyme is based on the position of 11β-HSD1 within the ER lumen where it colocalizes with hexose-6-phosphate dehydrogenase (H6PD). H6PD generates the reduced cosubstrate, NADPH. Thus, the ratio of NADPH/NADP confers directionality to 11β-HSD1 [18, 19]. Purified 11β-HSD1, in the absence of H6PD, behaves principally as a dehydrogenase, oxidizing cortisol to cortisone.
The ontogeny of 11β-HSD1 has been studied mainly in animal models and is predominantly expressed in the postnatal period. While 11β-HSD1 is detectable in many tissues, there is a lack of activity in early gestation with reductase activity only becoming apparent after delivery and rising steadily throughout infancy [20–22]. In humans, 11β-HSD1 ontogeny is less well characterized with few published studies. Cortisone therapy is ineffective in treating congenital adrenal hyperplasia in early infancy most likely due to absent or significantly reduced liver 11β-HSD1 [23]. Both reductase and dehydrogenase activity have been demonstrated in fetal lung tissue [24]. 11β-HSD1 activity remains similar throughout childhood in both boys and girls [25]. At puberty, there is a reduction in 11β-HSD1 activity in women which continues into adult life. In adults, there is a well-described dimorphism in cortisol metabolism between men and women with an apparent reduction in 11β-HSD1 activity in women [26, 27] although this is not consistent across all studies [28].
11β-HSD1 is expressed in many tissues including liver, adipose tissue, gonads, GI tract, kidney, eye, anterior pituitary, leukocytes, and bone [20]. Expression is highest in liver, brain, gonads, and adipose tissue. Many factors regulate expression and activity of 11β-HSD1. In most studies, GCs, proinflammatory cytokines (TNFα, IL-1β) peroxisome proliferator-activated receptor γ agonists, and CCAAT/enhancer-binding proteins (CEBPs) increase expression and/or activity. In contrast, growth hormone (GH) and liver X receptor (LXR) agonists decrease expression [20]. Recently, salicylates have been shown to downregulate 11β-HSD1 expression in adipose tissue and improve insulin sensitivity [29]. The effects of sex steroids, insulin, and other hormones are variable across tissues and between species. Estradiol has been shown to decrease 11β-HSD1 expression in rat liver and kidney, but testosterone was without effect [30].
Genetic defects in both HSDB1 and H6PD have been described. Cortisone reductase deficiency (CRD ) is caused by HSDB1 gene defects and apparent cortisone reductase deficiency (ACRD) by mutations in H6PD. Both cause a reduction in tissue 11β-HSD1 activity with low urinary cortisol metabolites, significantly elevated cortisone metabolites with a consequent compensatory increased HPA activity leading to hyperandrogenism, premature adrenarche, and PCOS in women, and precocious puberty in males [19, 31–35].
11β-Hydroxysteroid Dehydrogenase Type 2
In 1993, an enzyme with exclusive 11β-HSD dehydrogenase activity was identified from both human placenta and rat kidney [36], and in 1994, Krozoski et al. isolated human 11β-HSD from human kidney that was identical to the dehydrogenase enzyme found in the placenta [37]. This second enzyme was found to be distinct from 11β-HSD1 and was called 11β-HSD2 and is also a member of the SDR family.
The human HSD11B2 gene is located on chromosome 16, has 5 exons and is only 6 kb in length [38]. Human 11β-HSD2 contains 405 amino acids with a molecular weight of 44 kDa. It is also anchored to the ER and loses its dehydrogenase activity once dissociated from tissue membranes [37, 39]. 11β-HSD2 acts exclusively as a dehydrogenase across all species and has a Km for cortisol of 50–60 and 10–13 nM for cortisone [40]. Mutations in HSD11B2 lead to the syndrome of apparent mineralocorticoid excess (AME), a hereditary cause of life-threatening hypertension and hypokalemia, suppressed renin activity, and a metabolic alkalosis [41–45]. The underpinning mechanism relies upon the fact that cortisol is able to activate both the GR and mineralocorticoid receptor (MR) with equal affinity. Circulating cortisol concentrations far exceed those of aldosterone, the natural ligand for the MR, and therefore to prevent cortisol activating the MR in mineralocorticoid target tissues , 11β-HSD2 inactivates cortisol (to cortisone) locally. The condition is characterized by an increased urinary ratio of cortisol to cortisone metabolites. It can be treated with the synthetic GC, dexamethasone, which lacks mineralocorticoid activity, but is able to suppress endogenous cortisol production. Functional inhibition of 11β-HSD2 activity within the kidney is also the mechanism underpinning liquorice-induced hypertension [46].
11β-HSD2 is therefore expressed in aldosterone sensitive tissues , mainly in the distal nephron, colonic epithelium, salivary and sweat glands, and in the fetus and placenta during gestation. During gestation in humans and mammals, high levels of expression within the placenta protect the developing fetal tissues against excess GC exposure. Expression within the placenta steadily rises throughout gestation and declines two weeks prior to labor [22]. Altered or disrupted 11β-HSD2 activity, with subsequent excess intrauterine exposure to GC, has been implicated in “programming” effects upon the developing fetus leading to low birth weight and lifelong physiological consequences such as increased cardiovascular, metabolic, and psychiatric complications [47].
Unlike 11β-HSD1, there are considerable data published on the epigenetic influence on 11β-HSD2 activity in humans and in rodent models [48]. HSD11B2 is susceptible to epigenetic influence, with methylation of the promoter region of particular interest. Increased methylation of this region has been inversely associated with 11β-HSD2 expression and has been linked with the development of hypertension, intrauterine growth retardation, reduced birth weight, and neurobehavioral movement disorders [49]. In rodent models, intrauterine growth retardation has been associated with increased methylation of HSD11B2 gene promoter with subsequent repression of 11β-HSD2 expression in adult kidneys [50].
Factors that increase 11β-HSD1 expression tend to reduce 11β-HSD2 and include pro-inflammatory cytokines such as TNFα [51]. Estrogen increases 11β-HSD2 expression [30, 52]. Vasopressin has been shown to stimulate 11β-HSD2 [53]. Glucocorticoids downregulate 11β-HSD2 in fetal placenta and lung cells, but not fetal kidney [54, 55]. Hypoxia has also been shown to reduce 11β-HSD2 expression [56] whereas in colonic epithelium, aldosterone increases 11β-HSD2 expression [57].
A-Ring-Reductases
The A-ring reductases are important regulators of GC availability . 5αR1 and 2 have an important dual role in the prereceptor regulation of steroid hormone availability. They inactivate cortisol to dihydrocortisol which is then subsequently converted to tetrahydrocortisol through the activity of 3α-hydroxysteroid dehydrogenase (3α-HSD) and are therefore of crucial importance in local GC clearance. In addition to this role, 5αRs are fundamentally important in the reduction of testosterone to the more potent androgen, dihydrotestosterone (DHT), and therefore sit at an importance interface that sets the balance at a cellular level between GC and androgen action. Apart from GC and androgens, 5αRs can also metabolize other steroid substrates including progesterone and mineralocorticoids. 5αRs are microsomal enzymes and are NADPH dependent . While three isoforms of 5αRs have been identified to date [58, 59], with different biochemical properties and sensitivity to substrates, it is only type 1 and type 2 that appear to have a role in the regulation of steroid hormone availability.
5α-Reductase Type 1
The gene encoding 5αR1 (SRD5A1) lies on chromosomes 5 and has 5 exons and 4 introns. It consists of 259 amino acids with a molecular weight of 29 kDa. It is expressed in both human and mouse liver and also in skin (nongenital) and adipose tissue [58]. Although testosterone is the most widely recognized substrate of this enzyme, progesterone has a lower K m and therefore enzymatically may be the preferred substrate [60, 61]. To date, no mutations have been identified in SRD5A1.
Dutasteride is a dual inhibitor of both isoforms, 5αR1 and 5αR2, reducing circulating DHT by nearly 95 % compared to the baseline. MK-386 was reported to be a selective inhibitor of 5αR1 with 90 % efficiency but this compound is neither commercially available nor used in clinical practice [62].
5α-Reductase Type 2
The gene encoding 5αR2 (SRD5A2) lies on chromosomes 2 and has 5 exons and 4 introns. It has 254 amino acids with a molecular weight of 28 kDa and shares less than 50 % homology with 5αR1 [58, 63]. 5αR2 is expressed in human liver but not in mouse liver. 5αR2 is predominantly expressed in androgen-target organs such as prostate, epididymis, and seminal vesicles [58].
Finasteride is a selective 5αR2 inhibitor, while 5αR1 has a low sensitivity to this inhibitor. In comparison with 5αR1, 5αR2 has much higher affinity for androgen substrates such as testosterone. Many mutations and polymorphisms have been identified throughout the coding and noncoding regions of SRD5A2 [64]. Since 5αR2 converts testosterone to a more potent androgen (DHT), mutations in this enzyme lead to 46XY DSD (disorder of sex development) with consequent lack of virilization and poor development of the external genitalia. However, excessive androgen generation through the activity of 5αR2 has been implicated in conditions including polycystic ovary syndrome, breast cancer, and prostate cancer, as well as male pattern baldness [65–67].
5β-Reductase
The gene encoding 5β-reductase (or AKR1D1) is located on chromosome 7. 5β-Reductase is also highly expressed in hepatocytes and its crystal structure has been determined [68]. AKR1D1 is able to metabolize both cortisol and cortisone, and following 3α-HSD activity, it generates 5β-tetrahydrocortisol (5β-THF) and 5β-tetrahydrocortisone (5βTHE). While 5αRs reduce testosterone to the more potent 5α-DHT, 5β reductase generates 5β-DHT, which is inactive and thus limits androgen action locally. 5β-Reductase has a significant role in clearing the majority of all C-19-C21 steroids and therefore disruption of its activity has the potential to impact upon clearance of GCs, mineralocorticoids, and sex steroids. It also has an important role in bile acid production. Mutations in the gene encoding 5β-reductase lead to bile acid deficiency and form neonatal cholestatic liver disease which can progress to liver failure [69]. However, spontaneous recovery and survival into adult hood is reported [70].
Tissue-Specific Cortisol Metabolism
Adipose Tissue
In metabolic disease, alterations in adipose 11β-HSD1 in rodent models are well described. Activity is increased in visceral adipose tissue of obese, compared to lean, Zucker, and Wistar/obese (WNIN/ob) rats, and diabetic (db/db) mice [71, 72]. Additionally, in obese WNIN/ob and db/db but not Zucker diabetic fatty (ZDF) animals, 11β-HSD1 activity was increased in the subcutaneous depot [73, 74]. Interestingly, in Wistar rats short-term, but not long-term, high fat diet decreased 11β-HSD1 activity in subcutaneous and omental depots [75] suggesting an adaptive mechanism to protect against the short-term effects of high fat feeding.
11β-HSD1 knockout mice have an improved metabolic phenotype in comparison with wild-type littermates. They resist diet-induced obesity, have a more metabolically safe adipose distribution, gaining fat in the epididymal rather than the visceral depot, display improved glucose tolerance and insulin sensitivity, and have decreased circulating plasma fatty acids. Isolated adipocytes have increased insulin sensitivity [76, 77]. Transgenic mice overexpressing 11β-HSD1 specifically in adipocytes have a 15–30 % increase in adipose corticosterone (the predominant active GC in rodents) concentration and have increased food intake, and a small increase in subcutaneous and a dramatic increase in visceral adipose tissue mass [78]. These animals were also hypertensive, hyperglycemic, hyperinsulinemic, and glucose intolerant, with raised serum fatty acids and triglycerides [78, 79]. In a comparative study, a mouse overexpressing 11β-HSD2 in adipose tissue developed adipose tissue-specific GC deficiency. These mice had reduced fat mass and were resistant to weight gain on a high fat diet. Unexpectedly, the reduction in fat mass was predominantly due to a decrease in the subcutaneous depot, with a less dramatic upon visceral adipose. Globally, mice had improved glucose tolerance and insulin sensitivity; however, food intake was decreased and energy expenditure increased [80].
High levels of 11β-HSD1, but not 11β-HSD2, are expressed in human adipose tissue [81] where it functions largely as an oxoreductase, generating active GC and being induced by GCs and pro-inflammatory cytokines [82–84]. Whole tissue subcutaneous and omental adipose tissue depot expression levels are similar; however, H6PD and GR are more highly expressed in omental adipose tissue [85]. 11β-HSD1 expression is higher in omental compared to subcutaneous adipose stromal cells (contrasting with whole tissue expression data) and increases across adipocyte differentiation [86]. 11β-HSD1 inhibition blocks cortisone-induced differentiation [86, 87] and regulates GC-induced lipid accumulation [88].
Human expression studies have mainly focused on subcutaneous adipose tissue, and the majority of studies have shown that 11β-HSD1 expression and activity correlate positively with BMI and insulin resistance [89–96]. A few studies have examined omental adipose, and overall data suggest increased expression in obesity [85, 97–99]; however, this is not consistent across all studies [90, 100]. Stable isotope techniques have been used to demonstrate functional activity of 11β-HSD1 and while it is clear that adipose tissue is able to generate significant amounts of active GC, there is little evidence to suggest that intra-abdominal adipose actively ‘exports’ this to distant tissues [101]; however, active GC generated within subcutaneous adipose tissue can be exported to distant organs [101]. Importantly, in both intra-abdominal and subcutaneous depots there is shuttling between active and inactive GCs [102], thus altering the amount of locally derived GC, which in turn can have a potent impact upon adipose tissue biology.
11β-HSD2 expression has been described in human adipocytes although its true functional role has not been determined [103]. At a functional level, studies utilizing stable isotopes of cortisol that are able to distinguish oxoreductase versus dehydrogenase activity suggest that exclusive activity is the generation of cortisol within adipose tissue, as a result of 11β-HSD1 activity [104].
Liver
11β-HSD1 is expressed in rodent and human liver at high levels [14]. In rodent studies, hepatic 11β-HSD1 expression is decreased in some murine models of obesity [71, 73]. However, in the diabetic db/db mouse, hepatic 11β-HSD1 and GR expression are increased [108]. Global 11β-HSD1KO mice are protected from diet-induced hepatic steatosis [109] and, when fed a high fat diet, fasting glucose levels are significantly lower compared to controls [76]. In order to explore the role of hepatic 11β-HSD1 in global metabolic homeostasis, mouse models with liver-specific overexpression and knockdown have been developed. Transgenic mice overexpressing 11β-HSD1 under the hepatocyte-specific apoE promoter are hypertensive, dyslipidemic, and develop hepatic steatosis due to increased triglyceride accumulation and impaired lipid clearance. Interestingly, they do not develop steatohepatitis (NASH) and have only modest levels of insulin resistance when compared to adipose tissue-specific 11β-HSD1 overexpression [110]. Liver-specific 11β-HSD1KO mice have a mild metabolic phenotype, with a slight improvement in glucose tolerance (without significant improvement in insulin sensitivity) and no changes in hepatic lipid accumulation [111]. These data highlight the importance of extrahepatic 11β-HSD1 in regulating global and hepatic homeostasis.
In the human liver, 11β-HSD1 is localized centripetally with maximum expression around the central vein [112] and activity is exclusively oxoreductase [112, 113] generating active GC. In obese patients, the expression of GR, 11β-HSD1, and H6PD were all increased in the livers of patients with metabolic disease and were associated with disease severity [114]. However, in patients with proven non-alcoholic fatty liver disease (NAFLD), the expression of these genes was not altered [115, 116]. It is possible that 11β-HSD1 is differently regulated across the progression from steatosis to NASH. In patients with steatosis, total cortisol metabolites are increased, consistent with increased cortisol production yet hepatic 11β-HSD1 activity is decreased. However, in patients with NASH, activity was increased compared to controls and this might reflect the progression to a more inflammatory phenotype rather than simple lipid accumulation [117]. In patients with simple obesity, heaptic 11ß-HSD1 activity (as measured by cortisol generation form oral cortisone) is reducted [94, 118] as this is likely to largely (although not-exclusively [111]) reflect hepatic activity. However, stable isotope techniques have demonstrated preserved, rather than decreased, activity in patients with obesity and coexistent type 2 diabetes [119]. 11β-HSD2 is not expressed in the human liver.
There is an emerging role for the A-ring reductases in the prereceptor regulation of GC availability to modulate hepatic function. Rodent expression profiles differ from the human situation in that 5αR1 and not 5αR2 is expressed in rodent liver (both are expressed in humans). Rodent models have demonstrated that 5αR1 deletion is associated with increased hepatic steatosis as well as increased risk of progression to fibrosis and scarring in models of liver injury [120, 121]. As expected the changes were not seen in 5αR2 knockout models consistent with the lack of expression of 5αR2 in the normal rodent liver. 5β-Reductase is expressed in the rodent liver, but with the exception of its role in bile acid synthesis its contribution to other conditions using rodent models has not been explored.
Clinical studies have consistently demonstrated an association between worsening metabolic phenotype and increased 5αR activity as assessed most commonly by urinary steroid hormone metabolites [122–126]. In addition, patients with polycystic ovarian syndrome, which in itself is associated with insulin resistance and an adverse metabolic phenotype, have increased 5αR activity [125, 126]. Importantly, following aggressive weight loss in clinical studies 5αR activity decreases [124]. 5β-Reductase activity increases with hepatic lipid accumulation [127], but data on its role to regulate other aspects of metabolic pehnotype have not been explored.
Pancreatic Islet of Langerhans
There is continued debate about the localization and functional role of 11β-HSD1 in the pancreatic islet; studies have demonstrated colocalizations to the β-cell [169], while others have shown colocalization with glucagon in the periphery of murine and human islets, but not with insulin or stomatostatin, suggesting α- and not β-cell expression [128]. Several studies have demonstrated that pharmacological inhibition of 11β-HSD1 can regulate insulin secretion both in vitro [128–131] and in rodent models in vivo. Expression is increased in islets from obese ob/ob mice [129] and diabetic ZDF fa/fa rats, where 11β-HSD1 activity increased in proportion to hyperglycemia [132]. Prevention of hyperglycemia and hyperlipidemia by troglitazone, a PPAR gamma agonist, blocked the increase in 11β-HSD1; however, expression in isolated prediabetic islets was not altered by incubation with high glucose or oleate/palmitate, indicating that this was not a nutritional effect [132].
In a transgenic rodent model with β-cell-specific overexpression of 11β-HSD1, β-cell function was compromised with suppression of glucose-stimulated insulin secretion, but interestingly, in hemizygous mice fed there was reversal of β-cell failure on a high fat diet. This was thought to be due to an increased number and function of small islets, enhanced insulin secretion, and enhanced β-cell differentiation and survival. However, global 11β-HSD1 knockout mice have impaired β-cell function, with decreased glucose-stimulated insulin secretion [133]. Overall there remain many unanswered questions as to the role of 11β-HSD1 in the pancreatic islet and its true function is yet to be determined. 11β-HSD2 is expressed in whole islets although detailed localization and functional assessments have not been performed [134]. There are little if any data that have been published on the expression or activity of the A-ring reductase in the pancreatic islets. However, there does appear to be functional 5αR activity in fetal and pancreatic carcinoma tissue [135].
Skeletal Muscle
The role of 11β-HSD1 in skeletal muscle has not been examined in detail and the relative amount and activity in comparison with liver and adipose tissue is low, but oxoreductase activity has been demonstrated in human muscle explants, human primary cultures, murine explants, and transformed cell lines [136, 137]. Importantly, there are indications that skeletal muscle 11β-HSD1 activity may have a role in metabolic disease. Activity is increased in the gastrocnemius muscle of a rodent model of type 2 diabetes [138] and 11β-HSD1 inhibition increased skeletal muscle insulin receptor substrate 1 (IRS-1) mRNA expression and decreased expression of genes involved in lipid metabolism (lipolysis, lipogenesis, and lipid oxidation) [137]. Similar findings have been identified using human cell culture models [139], and in translational clinical studies, expression is increased in myotubes from obese type 2 diabetics, when compared to BMI-matched controls [140]. Increased expression is also associated with decreased grip strength with age [141, 142].
11β-HSD2, 5αR2, and 5β-reductase are not expressed to any significant level in skeletal muscle. 5αR1 however is expressed although its precise role is yet to be defined; however, inhibition of both 5αR1 and 2 using dutasteride was associated with decreased glucose disposal and this has been suggested to reflect a specific role of 5αR1 within skeletal muscle [105].
Cardiovascular System
11β-HSD1 and 2 are expressed in blood vessel walls and heart; however, oxoreductase directionality (11β-HSD1) predominates in vascular smooth muscle [143, 144]. 11β-HSD1 inhibition in apoE knockout mice achieved significant reduction atherosclerotic load suggesting a role in plaque formation [145]. Carbenoxolone (a nonspecific 11β-HSD1 and 2 inhibitor) treatment has been shown to reduce atherosclerosis in mice [146]. 11β-HSD1 in blood vessel epithelial cells may play a role in maintaining an antiangiogenic tone in vivo. In obesity, rapidly expanding adipose tissue becomes hypoxic, and this may drive inflammation, fibrosis, and insulin resistance. 11β-HSD1 knockout mice have enhanced vascularization and oxygenation of adipose tissue depots paralleled by increased expression of potent angiogenic factors including VEGF, apelin, and angiopoetin-like protein 4 [147]. Furthermore, 7 days after coronary artery ligation, 11ß-HSD1 knockout mice show increased vascularization in the infarcted myocardium, associated with partial protection against myocardial dysfunction [148].
11β-HSD2 is expressed in vascular endothelium [143]. 11β-HSD2 knockout mice develop endothelial dysfunction [149]. Lack of 11β-HSD2 and MR activation is implicated in generation of severe atherosclerosis in mouse models [150].
There is evidence linking 11β-HSD1 activity with atherosclerosis, and mediastinal adipose tissue 11β-HSD1 expression has been associated with coronary atherosclerosis [151]. The same authors demonstrated increased 11β-HSD1 expression in aortas of obese patients with the metabolic syndrome [152].
5αR1 is expressed in the vascular endothelium and smooth muscle. Most studies have evaluated its role in the context of functional inhibition or in the context of androgen administration. In rodent models, 5αR inhibition is associated with some endothelial damage and dysfunction [153], but currently data are lacking as to the contribution that cortisol clearance makes to these observations.
C entral Nervous System
11β-HSD1 is widely distributed in the adult brain, while 11β-HSD2 is only expressed at low levels. 11β-HSD1 is most highly expressed in the hippocampus, cortex, cerebellum, and anterior pituitary although expression is also found in the hypothalamus, amygdala, and brain stem. Additionally, expression and activity have been demonstrated in the choroid plexus and arachnoid granulation tissue of the brain ventricular system [154], as well as in the ciliary epithelium and trabecular meshwork of the eye [155]. Although 11β-HSD1 appears to act predominantly as an oxoreductase in the central nervous system (CNS) [156], its cofactor generating enzyme, H6PDH, does not universally colocalize with 11β-HSD1 and this has raised the suggestion that provision of NADPH to 11β-HSD1 in the CNS may not be exclusively related to H6PD [157].
A role for 11β-HSD1 in mediating memory loss and hippocampal atrophy is supported by data demonstrating that inhibition of 11β-HSD1 in cultured hippocampal cells reduced GC-induced neurotoxicity [156]. In aged mice and humans, 11β-HSD inhibition improves cognitive function, with similar results in aged 11β-HSD1KO mice [158–160]. However, in a recent study, selective 11β-HSD1 inhibition did not improve cognitive function in patients with Alzheimer’s disease [161].
In the ocular ciliary epithelium, 11β-HSD1 regulates aqueous humor production through increased local cortisol generation [155]. In a proof-of-principle study, the nonspecific 11β-HSD inhibitor, carbenoxolone, decreased intraocular pressure in patients with ocular hypertension [162].
Although the causal link is yet to be established, idiopathic intracranial hypertension (IHH) is associated with GC excess and also with simple obesity. In obese patients, dysregulation of 11β-HSD1 in the choroid plexus and arachnoid granulation tissue may be important in disease development. In obese subjects with IIH, global 11β-HSD1 activity decreases with weight loss and those with the greatest decrease in activity have the largest fall in intracranial pressure. In this study, weight loss was correlated inversely with CSF cortisone levels, suggesting decreased local 11β-HSD1 activity [163]. While the published data do suggest a role for 11β-HSD1 in the pathogenesis of IIH, proof-of-concept studies need to be undertaken using selective inhibitors in this group of patients.
11β-HSD2 is expressed at low levels in adult human brain. However, 11β-HSD2 is highly expressed in fetal (rat) brain [164] and has an important role in brain development. In normal anterior pituitary tissue, 11β-HSD2 mRNA is detected, but immunofluorescence has not been able to convincingly demonstrate protein expression. Interestingly, 11β-HSD2 expression is increased in ACTH-secreting corticotroph adenomas and therefore the consequent enhanced local inactivation of cortisol may explain, at last in part, their lack of response to circulating cortisol excess with resultant autonomous ACTH secretion [165].
While there is no doubt that the 5α-reduced steroids can impact brain function [166], it remains unclear as to how much of this impact is reliant upon their actions upon GCs. Similarly, 5β-reductase is expressed widely within the brain and reports have suggested that it is important for the local regulation of neuroactive steroid availability as well as potentially regulating extrahepatic bile acid synthesis that may function as neuroregulatory signaling molecules [167].
Inflammation and Immunity
GCs in pharmacological doses are immunosuppressive and produce powerful anti-inflammatory effects [168]. They achieve this by altering gene transcription and altering pro- and anti-inflammatory mediators including cytokines and signaling pathways. 11β-HSD1 is believed to play a key role in local inflammation and immune response to stimuli and allergens [169].
11β-HSD1 expression increases during monocyte to macrophage differentiation. In these cells expression is unaffected by pro-inflammatory cytokines, but is increased by IL-4, IL-13 and LPS [170]. Expression also increases when monocytes differentiate to dendritic cells under the influence of gm-CSF and IL-4. Activity is further increased by innate immune stimuli acting via toll-like receptors (TLRs), but is rapidly decreased by binding of the CD40 receptor, an adaptive immune stimulus [171]. Additionally, expression has been detected in murine CD4 and CD8 positive lymphocytes, B cells, and dendritic cells. Expression in CD4 positive lymphocytes increases with cellular activation or polarization into Th1 or Th2 cellular subsets [172].
11β-HSD1 knockout mice have defects in macrophage phagocytosis of apoptotic neutrophils during peritoneal inflammation [173]. In addition, they also display enhanced endotoxemia in response to LPS injection [174]. Furthermore, in models of joint inflammation, peritonitis, and lung inflammation, the inflammatory response was greater, and resolution slower, in 11β-HSD1 knockout mice [175], an observation that has raised concerns y about the clinical use of selective 11β-HSD1 inhibitors.
A limited number of studies have examined 11β-HSD1 in inflammation in humans in vivo. 11β-HSD1 activity is increased in patients with rheumatoid arthritis [176] and mRNA and protein levels are higher in biopsies of colonic tissue obtained from patients with colitis compared to control patient samples [177]. Additionally, acute exacerbations of inflammatory bowel disease are associated with a significant increase in systemic 11β-HSD1 activity, most likely originating from the inflamed bowel [178]. Interestingly, patients in remission also have high systemic activity, suggesting that local GC production within inflamed tissues might be sufficient to suppress the clinical features of inflammation. Taken together, these studies implicate 11β-HSD1 in having a role to limit the acute inflammatory response.
The response of the 5αR isoforms to inflammation is not fully defined. In a single study in patients with inflammatory arthritis, 5αR activity increased with anti-TNFα treatment although this was paralleled by a decrease in 11β-HSD1 [179]. Any potential role for 5β-reductase has noty been examined.
Bone and Joint
Osteosarcoma cells only express 11β-HSD2 and this contrasts with primary osteoblasts which exclusively express 11β-HSD1 [180, 181]. Ex vivo assays using bone chips have shown bidirectional interconversion of cortisone and cortisol, with kinetics suggesting 11β-HSD1 rather than 11β-HSD2 activity [182]. Although expression appears primarily localized to osteoblasts, some expression is also seen in osteoclasts in human adult bone. Expression of 11ß-HSD1 is regualted across osteoblast differentiation and cortisone treatment of cells in culture enahnces cellular differentiation [183]. Activity of 11β-HSD1 in osteoblasts is increased by both pro-inflammatory cytokines and glucocorticoids [184] in a synergistic fashion [185], mediated via the nuclear factor-kB (NF-kB) and p38 mitogen-activated protein kinases (MAPK) pathways.
In global 11β-HSD1 knockout mice there are no changes in commonly measured parameters of bone mass and geometry [186]; however, increased circulating levels of corticosterone in this model limit the significance of these findings. Additionally, the phenotype has only been examined in young mice and it is likely that any bone phenotype would be most evident in older animals. Interestingly, targeted overexpression of 11β-HSD2 within osteoblasts, resulting in cell specific GC deficiency, causes subtle abnormalities of skeletal structure including reduced vertebral size and density and reduced cortical width [187].
The presence of 11β-HSD1 in bone raises the possibility that its activity may predict clinical susceptibility to GC-induced osteoporosis. In healthy subjects, the ratio of urinary cortisol to cortisone metabolites predicts the response of bone formation markers to prednisolone treatment [188].
The role of 5α-reductase isoforms has begun to be explored in rodent models although not in humans at present. 5αR1 knockout mice have a sexually dimorphic phenotype with decreased bone mineral content and bone density in male mice with increased bone mass in female mice. The authors postulate that this reflects local changes in androgen availability, but the contribution of alteration in tissue-specific GC concentrations was not assessed [189].
Skin and Salivary Glands
Cortisol metabolism within skin is rapidly becoming an area of interest and investigation. Skin has been shown to be an active site of cortisol production and metabolism [190, 191]. Excess skin exposure to GCs causes skin changes similar to the natural aging process including reduced elasticity, reduced collagen and fibroblast numbers, thinning of dermis, and epidermis and a reduction in the repair capacity of skin. Increased exposure to GCs has been postulated as a factor in age-related changes, inflammatory, and autoimmunity changes seen in skin [192]. It has been postulated that skin changes seen over time are in part a result of 11β-HSD1 activity [190].
Both 11β-HSD1 and 2 are expressed in skin [190, 191, 193]. 11β-HSD2 is expressed in association with the mineralocorticoid receptor on sweat glands; however, its role (if any) within the dermis and epidermis is debated. In wound healing, 11β-HSD2 expression has been shown to be induced 48 h after tissue injury with subsequent return to basal levels at 96 h [191]. This has been postulated to be a mechanism to reduce local cortisol excess following inflammation.
11β-HSD1 is widely expressed in human and mouse dermis and epidermis [190, 193, 194]. Upon differentiation of keratinocytes 11β-HSD1 expression increases [193], somewhat akin to the changes seen with preadipocyte differentiation [195]. Interestingly despite reducing levels of expression of 11β-HSD1 in elderly subjects, a paradoxical rise in 11β-HSD1 activity is seen with increasing age in both humans and mice [190]. This gives credence to the concept of age related skin atrophic changes being in part due to increased cortisol exposure secondary to increased 11β-HSD1 activity.
11β-HSD1 has been shown to have a pivotal role in skin repair following injury and tissue remodeling [193, 196, 197]. In mice, 11β-HSD1 contributes to impaired wound healing. Blocking 11β-HSD1 enhances wound healing in mice and prevents age-induced skin changes [196]. These data suggest that local cortisol, generated by 11β-HSD1, is critically important in wound healing and in aging skin changes. Inhibitors of 11β-HSD1 (topical or oral) may therefore have therapeutic potential.
As mineralocorticoid target tissues, both skin and salivary glands express 11β-HSD2. In the skin expression is mainly restricted to sweat glands [198]. 11β-HSD2 is expressed in both parotid and submandibular glands [198, 199] and measuring salivary cortisone has been postulated a potential biomarker of serum-free cortisol [200]. In addition, reduced activity of 11β-HSD2 in sweat glands has also been linked with essential hypertension [201].
The role of the A-ring reductases in skin has been extensively examined in the context of androgen generation and in particular its relationship to the development of hirsutism and the potential for local generation of DHT. Their role in cortisol metabolism within the skin has not been determined.
Kidney
11β-HSD2 is the predominant isoform in the human kidney, although 11β-HSD1 is expressed in the rodent kidney. The role of 11β-HSD2 in the kidney is to protect the MR from excess exposure to GC. 11β-HSD2 is widely expressed in distal nephrons [39]. Although the inherent enzyme ability of 11β-HSD2 to clear cortisol (converting it to cortisone) should not be enough, given concentrations and binding affinities, in reality it protects the mineralocorticoid receptor from GC exposure [202]. Lack of 11β-HSD2 in kidney leads to life-threatening hypertension and hypokalemia. Reduced 11β-HSD2 activity, as measured by urinary steroid metabolites ratios, has been associated with essential hypertension in aging populations [203] as well as in those with underlying renal impairment [204, 205].
Colon
11β-HSD2 is expressed in colonic epithelium [207]. Expression is increased by aldosterone in rats [57]. In Inflammatory bowel disease 11β-HSD2 expression is downregulated in both humans and rats [177]. This is accompanied by an increase in 11β-HSD1 expression and so is presumed to be an attempt to locally control GC exposure to inflamed tissue. This has been discussed in the section on immunity and inflammation above. Zhang et al. showed that inhibiting 11β-HSD2 reduces colon carcinogenesis by inhibiting COX 2 pathways. The reduction in 11β-HSD2 blocked colorectal adenocarcinoma angiogenesis and metastasis [208]. There are currently no data with regards to the role of the A-ring reductase and GCs within the colon.
Pharmacological Targeting of Prereceptor GC Metabolism
11β-HSD1 Inhibition
While the clinical consequences of 11β-HSD2 inhibition (as exemplified by liquorice consumption) are detrimental, over the last 10–15 years, there has been a significant drive to develop selective 11β-HSD1 inhibitors based upon the premise that decreasing tissue-specific cortisol availability, notably in the context of metabolic disease, is likely to have a beneficial impact.
Carbenoxolone is a nonselective 11β-HSD inhibitor . In healthy individuals, it improves whole body insulin sensitivity [209], and in patients with type 2 diabetes, it decreases glucose production rates, principally through a reduction in glycogenolysis with no apparent effect on gluconeogenesis. In addition, it decreases total circulating cholesterol levels [210]. Its beneficial effects are modest, and while this is most likely to reflect its nonselective action, questions have arisen as to its ability to access key metabolic target tissues, including adipose, although studies have shown therapeutic levels within adipose interstitial fluid [96, 211]. Carbenoxolone has also been shown to impact upon bone biology in vivo . In a proof-of-principle study there were no changes in bone formation markers, but bone resorption decreased significantly [182].
Several phase II studies have now been published that have examined selective 11β-HSD1 inhibitors. INCB013739, when administered to patients with type 2 diabetes twice daily for 2 weeks, completely abolished all conversion of oral cortisone to cortisol. Metabolically hepatic glucose production rates decreased, without alteration in glucose disposal. Interestingly, the decrease in fasting glucose was most marked in the most hyperglycemic patients. In addition, total and LDL cholesterol decreased with no change in HDL-cholesterol or triglyceride levels. In a double-blind placebo-controlled study, patients with type 2 diabetes with inadequate glycemic control on metformin therapy (HbA1c 7–11 %) were randomized to receive 5, 15, 50, 100, or 200 mg INCB13739 in addition to metformin for 12 weeks. Weight, glycemic control, and lipid profile all improved although the effects were relatively modest with reductions in HbA1c of approximately 0.5 % and a small reduction in HOMA-IR consistent with insulin sensitization. As expected, treatment with this class of agent activates the HPA axis (as a consequence of decreased cortisol half-life) with consequent elevation of adrenal androgen secretion. There were no changes in HDL or free fatty acids and blood pressure was not affected [212].
Data have also been published on additional compounds; MK0916 was given to patients with type 2 diabetes. While it was well tolerated, MK0916 had only very modest effects on metabolic parameters . There was a decrease in weight and waist hip ratio in the 6-mg group and in this group there was also a small reduction in HbA1c (0.3 %); however, no change was seen in fasting plasma glucose, 2 h postprandial glucose, or fasting or postprandial serum insulin [213]. A further compound, MK0736, has also been tested in obese and overweight hypertensive patients. Both doses of the compound tested decreased blood pressure. Again, consistent with other studies all active treatments caused a small but significant decrease in weight [214].
PF-915275 is an effective 11β-HSD1 inhibitor as measured by changes in urinary steroid metabolite ratios and prednisone to prednisolone conversion, but to date there are no data on the impact of this compound on metabolic phenotype [215]. Most recently, RO5093151 has been trialed in the context of hepatic steatosis. The drug appeared safe and well tolerated and did reduce hepatic steatosis as measured by magnetic resonance spectroscopy although in absolute terms the reduction was once again modest, but the duration of the study was only 12 weeks [216].
5α-Reductase Inhibition
Clinical studies have highlighted the potential role for 5α-reductase in the regulation of metabolic phenotype, although there is still debate as to whether the abnormalities observed represent the cause or consequence of disease. As described above, cross-sectional and longitudinal studies have demonstrated increased 5α-reductase activity with insulin resistance and increasing adiposity and reductions following weight loss [122–124]. A recently published study has examined the metabolic impact of selective 5αR inhibition in humans [105]. Following a 3-month treatment period, the authors observed inhibition of glucose disposal under hyperinsulinemic conditions with dutasteride treatment (nonselective 5αR1 and 2 inhibitor) but not finasteride (selective 5αR2 inhibitor), which may reflect the impact of inhibition of 5αR1 activity within skeletal muscle. The long-term clinical consequences of these observations remain to be determined as well as the identification of the mechanisms responsible, in particular their dependence upon either GC and/or androgen metabolism.
Conclusion
GCs have multiple actions across almost all tissues in the body and the regulation of their action is complex. Prereceptor GC metabolism, either regeneration of active cortisol through the activity of 11β-HSD1 or clearance via 11β-HSD2, and the A-ring reductase are potently able to impact upon GR activation. The consequences of their activity not only are dependent upon the precise pattern of expression within specific tissues but also may reflect the broad range of substrates (including GCs) that they are able to metabolize. Dysregulated expression has been implicated in the pathogenesis of many diseases, and the fundamental importance of the prereceptor GC concept is highlighted by patients with genetic defects that are potentially life-threatening. Pharmacological intervention, specifically targeting 11β-HSD1, has progressed all the way through to phase II clinical trials and while the outcomes with respect to metabolic disease have been positive, the magnitude of the response has perhaps been less than had been anticipated. This may reflect targeting of therapy to specific tissues but also the fact that only the ‘regenerated’ part of GC has been blocked. In terms of the future, the role of 11β-HSD1 in the skin and its involvement in wound healing make it an attractive therapeutic prospect. In addition, there is emerging evidence that 11β-HSD1 may have a role in the regulation of tissue-specific exposure to exogenously administered GCs, raising the possibility that 11β-HSD1 inhibitors could have utility in reducing the adverse effects of prescribed GCs [217, 218].
References
1.
Munck A, Naray-Fejes-Toth A. The ups and downs of glucocorticoid physiology. Permissive and suppressive effects revisited. Mol Cell Endocrinol. 1992;90(1):C1–4.PubMed
2.
Esteban NV, Loughlin T, Yergey AL, Zawadzki JK, Booth JD, Winterer JC, et al. Daily cortisol production rate in man determined by stable isotope dilution/mass spectrometry. J Clin Endocrinol Metab. 1991;72(1):39–45.PubMed
3.
Siiteri PK, Murai JT, Hammond GL, Nisker JA, Raymoure WJ, Kuhn RW. The serum transport of steroid hormones. Recent Prog Horm Res. 1982;38:457–510.PubMed
4.
Keenan DM, Roelfsema F, Veldhuis JD. Endogenous ACTH concentration-dependent drive of pulsatile cortisol secretion in the human. Am J Physiol Endocrinol Metab. 2004;287(4):E652–61.PubMed
Related posts:
 and Treatment of Aggressive Corticotroph Pituitary Tumors
Regulation of Body Composition and Metabolism
Regulation of Neurocognitive and Neuropsychiatric Function
Strategies for Localization of ACTH-Secreting Tumors
Therapies in Cushing’s Syndrome
and Treatment of Aggressive Corticotroph Pituitary Tumors
Regulation of Body Composition and Metabolism
Regulation of Neurocognitive and Neuropsychiatric Function
Strategies for Localization of ACTH-Secreting Tumors
Therapies in Cushing’s Syndrome
 and Nonneoplastic/Physiological Hypercortisolism: Cushing Versus Pseudo-Cushing Syndromes
and Nonneoplastic/Physiological Hypercortisolism: Cushing Versus Pseudo-Cushing Syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


