Medullary Thyroid Carcinoma
Mimi I. Hu
Camilo Jimenez
Gilbert J. Cote
Robert F. Gagel
Introduction
Medullary thyroid carcinoma (MTC) is an uncommon malignant tumor derived from the C or calcitonin producing cells of the thyroid gland. It is notable for being one of the last forms of thyroid carcinoma to be identified. The first description by Hazard in 1959 separated this form of aggressive thyroid carcinoma from other poorly differentiated or anaplastic forms of thyroid carcinoma (1). This description of MTC presaged three periods of intense investigation over a half-century that has brought our understanding of the pathogenesis of this unusual thyroid cancer to a remarkable level and has resulted in ground-breaking targeted therapy for patients with metastatic cancer.
The first period occurred during the 1960s and early 1970s, initiated by a report by Sipple (2) describing the association of MTC and pheochromocytoma in a single patient and describing several other examples from the literature, an association now known as multiple endocrine neoplasia type 2A (MEN2A). Although Sipple did not correctly identify the thyroid cancer as MTC, Williams (3) and others (4,5) quickly pieced together the association of MTC and pheochromocytoma and correctly hypothesized that MTC was derived from the parafollicular or C cell, a cell type that produced the newly discovered peptide, calcitonin (CT) (6,7,8). Over the course of the next 3 to 5 years several groups demonstrated the correctness of this hypothesis by demonstrating that MTC produces calcitonin (9,10) and that measurement of CT could be used to diagnose MTC (11).
A second intense period of discovery surrounded the mapping of the gene for MEN2 and the discovery that mutations of the RET proto-oncogene (12,13), a tyrosine kinase receptor (14), caused not only this genetic syndrome but also were found as somatic mutations in approximately 25% of sporadic MTCs (15). The recognition of RET as the major cause of MTC led to the identification of all the major components of the RET receptor system, an understanding of the role of the receptor system in normal developmental biology of the sympathetic nervous system, and the beginnings of an understanding of how mutations of RET cause transformation. More importantly these discoveries created a new paradigm for management of genetic malignancy: Identification of gene carriers and removal of the organ containing cells at risk for transformation early in life.
The remainder of this chapter will chronicle these remarkable events and, more importantly, attempt to place these discoveries into a context of clinical usefulness.
The Thyroidal C cell
The C or calcitonin producing cell is a neuroendocrine cell representing less than 1% of total cells within the thyroid gland. The C cells are distinct and separable from the more ubiquitous thyroid follicular cell. Classical studies by Le Douarin demonstrated migration of cells from the neural crest and differentiation into C cells of the ultimobranchial body, a discrete and separable entity in the neck that is the repository of C cells in birds and fish (16,17). Other more recent studies using genetically modified cells indicate that mammalian C cells are derived from the endodermal epithelial cells of the fourth pharyngeal pouch (18). In mammalian species these cells migrate into the thyroid gland during development and occupy a characteristic central location at the junction of the upper one-third and lower two-thirds of each thyroid lobe. This developmental pattern is of significance because it explains the characteristic location of hereditary MTC (Fig. 51.1).
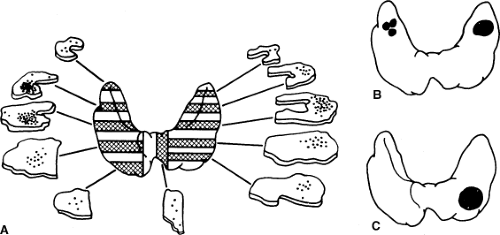 Figure 51.1 Characteristic location of medullary thyroid carcinoma in hereditary and sporadic types. A: C cells are found at the junction of the upper one-third and lower two-thirds of each thyroid lobe. B: In hereditary MTC, each C cell expresses a mutant RET proto-oncogene receptor leading to multicentric MTC. Hereditary MTC is usually bilateral and most commonly located at the junction of the upper one-third and lower two-thirds of each lobe of the thyroid gland, the location of the greatest concentration of C cells. C: Sporadic MTC is thought to derive from transformation of a single cell. |
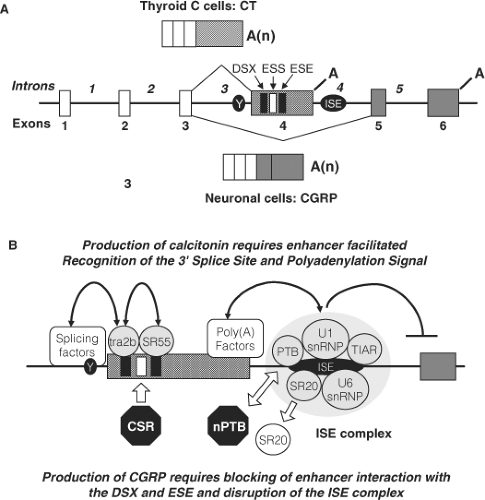 Figure 51.2 Alternative Splicing of the Calcitonin/CGRP (CALCA) Gene. (A) Transcripts derived from the six exon CALCA gene are alternatively spliced to produce mRNAs encoding either calcitonin (CT) or calcitonin gene-related peptide (CGRP). In thyroid C cells CT is produced by RNA splicing to include exon 4; in neuronal cells CGRP is produced by skipping this exon. Studies of the rat and human genes have identified five key sequences that are involved in recognition of exon 4. These sequences include: a pyrimidine branch point (Y), which creates a “weak” 3′ splice site, a doublesex repeat element (DSX), an exonic splicing silencer (ESS), an exonic splicing enhancer (ESE) and an intronic splicing enhancer (ISE). (B) The production of CT mRNA requires facilitated recognition of exon 4. The binding of tra2b to the DSX and SRp55 to the ESE are thought to facilitate recognition of the 3′ splice site by splicing factors. The downstream ISE element forms a large complex that binds several factors. The binding of polypyrimidine binding protein (PTB), SRp20, and U1 snRNP are thought to stimulate polyadenylation factors, while binding of U6 snRNP and TIAR are proposed to inhibit recognition of the exon 5 splice site. The production of CGRP requires the blocking or disruption of enhancer complexes. A brain-specific Candidate Splicing Repressor (CSR) has been demonstrated to bind the ESS sequence to prevent the binding of tra2b and SRp55 as discussed in the text. In addition the formation of the ISE complex is disrupted in neural cells due to the presence of a neural-specific PTB (nPTB) and reduced levels of SRp20. In the absence of facilitated recognition, exon 4 skipping is thought to be the default pathway. |
The C cell is a neuroendocrine cell with quasi-neuronal properties (uptake of biogenic amines and neuronal features when placed in tissue culture) combined with endocrine secretory peptide production analogous to adrenal chromaffin, pancreatic or gastrointestinal neuroendocrine, and certain pituitary cells. C cells secrete calcitonin and other small peptides (somatostatin) directly into blood vessels; this contrasts with deposition of the secretory products of the thyroid follicular cell into the thyroid follicle. In a previous era, members of this class of neuroendocrine cells were called APUD cells (amine precursor uptake and decarboxylation), (19) and described a complex and multi-organ class of neuroendocrine cells; this classification has fallen out of favor as the differences between members of this group of cells have been elucidated.
Features that make the C cell unique
C cells, be they located in the ultimobranchial body (birds and fish) or distributed throughout the thyroid gland (mammals), are characterized by the production of calcitonin. This small 32 amino acid peptide lowers the serum calcium concentration when injected into a rodent (6,8). These effects are mediated through binding of calcitonin to a specific receptor (the calcitonin receptor or CTR) on the osteoclast (20), a specialized cell that resorbs bone, causing it to retract and cease bone resorption (21). More recent studies suggest a more complicated role for CT in bone remodeling. Deletion of the calcitonin/calcitonin gene-related peptide CT/CGRP gene in a mouse model leads to a higher bone mass, (22) a finding that is supported by a similar phenotype in mice in which the CT receptor is deleted (23) In both model systems, disruption of the CT signaling pathway leads to increased bone formation, a surprising finding that argues for a role of this peptide as an inhibitor of bone formation. Thus the major physiologic effects of CT appear to be the short-term inhibition of bone resorption,
perhaps to protect against acute increases in the plasma calcium concentration, and a subtle and potentially more important long-term effect to regulate bone formation.
perhaps to protect against acute increases in the plasma calcium concentration, and a subtle and potentially more important long-term effect to regulate bone formation.
Although calcitonin is produced in multiple neuroendocrine cell types in mammalian species (neuroendocrine cells in the adrenal, pancreas, lung, prostate, breast, etc. and in any cell type during sepsis (24,25), under normal physiologic conditions the thyroidal C cell is the predominant source of CT. Removal of the thyroid gland in normal mammalian species lowers plasma concentrations of CT to nearly undetectable levels. A point of some significance in the clinical management of MTC is that during infection or inflammation CT can be produced in multiple cell types, a point that may explain the persistent elevations of plasma CT observed for several months following thyroidectomy (26).
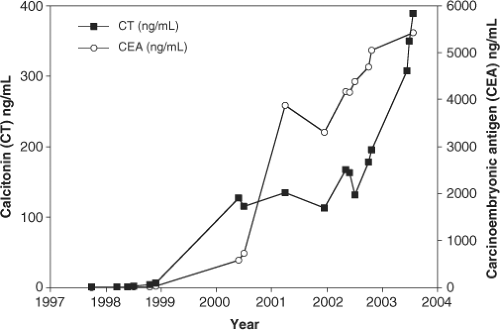 Figure 51.3 Measurement of serum calcitonin (CT) and carcinoembryonic antigen (CEA) in a patient with progressive medullary thyroid carcinoma (MTC). Serum CT and CT rose progressively in this patient with sporadic MTC. Note the point-to-point variability of CT and CEA, although the trend over the 5 to 6 years of observation is clear. The patient died from hepatic failure caused by metastatic MTC shortly following the last measurements. |
The calcitonin/calcitonin gene-related peptide (CT/CGRP) gene (CALCA) contains 6 exons (Fig. 51.2). Alternative RNA processing of the primary transcript by inclusion of exons 1 to 4 with polyadenylation following exon 4 (encoding calcitonin) produces a mRNA that encodes the precursor for calcitonin; processing the same transcript to exclude exon 4 produces a mRNA that encodes the precursor for CGRP. The processing of the primary transcript has been studied extensively. The splice signals surrounding exon 4 are weak, particularly an atypical pyrimidine branch point instead of the usual adenine, making inclusion of exon 4 in the final transcript an unfavored event. This suggests that exon 4 exclusion is the default pathway. Therefore, the inclusion of this exon to produce calcitonin requires a mechanism that enhances the recognition of the exon 4 splicing and polyadenylation sequences and is
ubiquitous. Selective interaction of transacting factors with repeating sequences within exon 4 by a tra-like protein and Srp55 (27) enhance recognition of the splice site, while a large complex of factors bound to a novel intronic sequence downstream of the exon 4 polyadenylation signal enhances polyadenylation and simultaneously inhibits splicing to the CGRP exon (28,29). These regulatory events lead to selective inclusion of exon 4 in C cells and production of CT. The mechanisms involved in the production of CGRP are less well studied, but are thought to involve the disruption of enhancer complexes (29,30). In MTC there is disordered RNA processing that causes production of a greater percentage of CGRP than normal; at present there is no evidence that the disordered splicing plays a pathogenetic role in transformation of the C cell.
ubiquitous. Selective interaction of transacting factors with repeating sequences within exon 4 by a tra-like protein and Srp55 (27) enhance recognition of the splice site, while a large complex of factors bound to a novel intronic sequence downstream of the exon 4 polyadenylation signal enhances polyadenylation and simultaneously inhibits splicing to the CGRP exon (28,29). These regulatory events lead to selective inclusion of exon 4 in C cells and production of CT. The mechanisms involved in the production of CGRP are less well studied, but are thought to involve the disruption of enhancer complexes (29,30). In MTC there is disordered RNA processing that causes production of a greater percentage of CGRP than normal; at present there is no evidence that the disordered splicing plays a pathogenetic role in transformation of the C cell.
Calcitonin gene expression is enhanced by activators of protein kinase A (31), protein kinase C (32), and mitogen-activated protein (MAP) kinases (33) It is inhibited by treatment with 1,25 dihydroxyvitamin D3 (34,35), retinoic acid analogues (36), nerve growth factor (37 and 5-hydroxytrytamine agonists (38). Glucocorticoids have complex effects on transcription (39,40,41). Tissue-specific expression of the calcitonin gene is regulated by two widely separated enhancer regions. The first (distal enhancer) is located 1000 nucleotides (42,43) and the second (proximal enhancer) approximately 250 nucleotides (31) upstream of the transcription start site. The distal enhancer is composed of three functional CANNTG or E-box motifs that bind helix-loop-helix (HLH) transcription factors (42). Members of the USF-1 and USF-2 HLH family of proteins, in combination with other undefined proteins, constitute the major transcription complex regulating the distal enhancer and likely confer tissue specificity (44). The proximal site contains cAMP response elements (CRE) and an overlapping octamer homeodomain-like binding site (CREL/O). These sequences mediate the cAMP/protein kinase A responsiveness of the cell (31). MAP kinase activation of CT/CGRP gene transcription is mediated through a Ras interacting site near the CRE that binds a novel and as yet unidentified zinc-fingered protein (33). The active metabolite of vitamin D, 1, 25 dihydroxyvitamin D3, inhibits transcription of the calcitonin gene by interfering with interaction between transcription complexes that occupy the upstream and downstream enhancers (35).
Calcitonin secretion is regulated primarily by the extracellular calcium concentration. An increase in the serum calcium concentration above normal physiologic levels activates the calcium sensing receptor (CaSR) (45,46), a G-protein coupled receptor that facilitates increased calcitonin secretion. Binding of CT to its receptor on the osteoclast inhibits bone resorption (47). The resulting reduction in calcium concentration leads to lowered CT secretion. Other stimuli that enhance calcitonin secretion include supraphysiologic concentrations of glucagon (48), 5HT1 receptor agonists (49) gastrin (and pentagastrin) (50), alcohol (51), and exercise (52) and glucagon-like peptide-1 (GLP-1) (53) A detailed discussion of each of these stimuli is beyond the limits of this chapter and the reader is referred to a review on this subject for more detail (54) The finding that glucagon-like peptide-1 (GLP-1) agonists, an effective therapy for type 2 diabetes mellitus, bind to GLP-1 receptors, stimulates CT release and cause MTCs in rodents has stimulated a broad discussion regarding their safety in humans (53). This will be discussed in a section below.
The recognition that 90% of CT is derived from the thyroid (except during periods of inflammation or sepsis) and that serum levels of CT are elevated in patients with MTC has made it a useful tumor marker. The sensitivity of CT measurement was further enhanced by combining it with calcium (11) and/or pentagastrin (50) stimulation and formed the basis for early identification of this tumor in patients with hereditary MTC. Calcitonin measurements are used less frequently for early diagnosis of MTC today, largely replaced by genetic tests for diagnosis of hereditary MTC.
Calcitonin measurements are also used to follow patients with residual or metastatic MTC. Sequential measurements of calcitonin over extended periods of time are useful for quantitating tumor mass; there is an almost direct correlation between CT measurements and tumor volume (Fig. 51.3). There are several points to keep in mind regarding CT measurements. The first is that CT is a secretory peptide whose secretion is episodic and may be affected by the ambient plasma calcium concentration, exercise, eating (gastrin stimulation), or other poorly defined stimuli. Calcitonin measurements may normally vary by a factor of 2- to 3-fold and therefore direct comparison of any two CT measurements provides little useful information. Aggressive and dedifferentiated MTCs, paradoxically, may lose
the ability to transcribe the CT/CGRP gene and thus a falling CT value may actually be an indicator of a poor prognosis (55). Another pragmatic observation: patients with serum CT values >5,000 pg/mL (normal being <10 pg/mL in most assays) following total thyroidectomy and lymph node dissection most commonly have metastatic disease outside the neck. Neck and mediastinal disease in such a patient is almost always detected by ultrasound or high quality CT scan of the chest or abdomen; in cases where these radiographic studies are negative, the most likely site of metastasis is the liver where microscopic metastases are difficult to detect.
the ability to transcribe the CT/CGRP gene and thus a falling CT value may actually be an indicator of a poor prognosis (55). Another pragmatic observation: patients with serum CT values >5,000 pg/mL (normal being <10 pg/mL in most assays) following total thyroidectomy and lymph node dissection most commonly have metastatic disease outside the neck. Neck and mediastinal disease in such a patient is almost always detected by ultrasound or high quality CT scan of the chest or abdomen; in cases where these radiographic studies are negative, the most likely site of metastasis is the liver where microscopic metastases are difficult to detect.
Carcinoembryonic antigen (CEA) is a useful tumor marker for monitoring the growth rate of MTC (56,57). There is less short-term variability of plasma concentrations than observed for CT, making this peptide a useful marker for monitoring tumor mass (Fig. 51.3). Since CEA is made throughout the GI track and liver and may be elevated in cigarette smokers and in several other tumor types, there is a broad range of normality, making it an insensitive and inaccurate marker for detection of early MTC.
However, both serum CT and CEA are useful markers for assessing disease progression over time. There is a direct correlation between the rate of increase of these tumor markers and progression of disease. Patients with a serum CT or CEA doubling time of less than two years are most likely to develop progressive metastatic disease and die from their disease, whereas patients with longer doubling times progress much more slowly or may not progress at all (58).
Other genes that are expressed by normal C cells and may be produced by MTC include somatostatin (59), histaminase (60), and chromogranin A (61); none are specific for MTC. MTC may also express a number of other genes that are not normally expressed by the C cell or are expressed at low levels. These include proopopiomelancortin (POMC) (62), vasoactive intestinal peptide (VIP), and neurotensin (63), and bombesin (64). Approximately 5% of ectopic ACTH (adrenocorticotropic hormone) syndrome is caused by MTC, due to tumor production of ACTH (65).
Histologic Features of MTC
MTC develops as a firm, white, almost chalky tumor within the thyroid gland. The transformed C cells are usually polyhedral or polygonal in shape and may be arranged in a variety of patterns (1,66). Amyloid distributed throughout the tumor is common in slowly growing tumors (67) whereas cellular components predominate in more rapidly growing tumors. Tumor calcification, related to deposition of calcium in the amyloid, is found commonly and may be detected by x-ray.
The combination of amyloid and immunohistochemical staining for CT form the most characteristic features of MTC (68). In rare, poorly differentiated MTCs the ability to produce CT is lost, a finding that may indicates a poor prognosis. Other tumors that produce CT and should be considered in the differential diagnosis include breast, prostate, and small cell lung carcinoma and other neuroendocrine tumors including pheochromocytoma or paraganglioma, islet cell carcinoma, and carcinoid (68). The diagnosis of MTC can be made easily by fine needle aspiration, combining CT immunohistochemical staining and the presence of amyloid (69); none of the other neuroendocrine tumors have this characteristic constellation of findings.
Clinical Presentation of MTC
One of the fascinating features of this tumor is the variety of clinical presentation. The most common is as a single or multiple cold thyroid nodules, on radioactive scan with or without palpable lymph nodes. This tumor may also present in the context of a known kindred with hereditary MTC. In most such examples, early screening by genetic testing will result in a thyroidectomy prior to the presence of any detectable disease. In rare examples of hereditary MTC, to be discussed later in this chapter, patients may present with clinical features of a pheochromocytoma (MEN2A or MEN2B), clinical features of Hirschsprung disease with intestinal obstruction or pseudo-obstruction (MEN2A or MEN2B), or hyperparathyroidism. A rare presentation is the development of a pruritic cutaneous lesion over the upper back known as cutaneous lichen amyloidosis that is seen in the context of MEN2A (70,71) A small percentage of patients, generally with widespread disease and hepatic metastasis, may present with diarrhea. It is not uncommon for such a patient to bounce from subspecialist to subspecialist before the correct diagnosis is achieved (72,73,74). Perhaps the most unusual example one of the authors has observed is a patient with breast cancer with persistent elevation of CEA for years, presumed to have metastatic breast cancer, who presented with diarrhea some years later and was belatedly discovered to have MTC, a presentation similar to that reported earlier (75). Finally, the increasing use of tumor marker measurement leads to the inevitable finding of a patient with an unexplained elevation of CEA or CT. Each of these clinical presentations will be woven into the subsequent sections discussing the specific clinical syndromes associated with MTC.
Clinical Syndromes Associated with MTC
Sporadic MTC
Sporadic MTC is most common form of this tumor. It is thought to arise, de novo, as a result of a somatic mutation(s) of the RET proto-oncogene or other genes in a single parafollicular or C cell. Sporadic MTC is most commonly unicentric, although multicentric disease may occur in some cases. Six to 7% of patients with apparently sporadic MTCs are subsequently found to have a germline RET mutation indicative of hereditary disease (discussed in detail in a later section) (15).
An isolated thyroid nodule is the most common presentation. The parafollicular cell does not concentrate iodide and therefore radioiodine scans (131I or 123I) show a “cold” thyroid region relative to surrounding thyroid tissue. The diagnosis is most commonly made by fine needle aspiration and confirmed by the measurement of a serum CT and/or CEA. Thyroid cytopathology for MTC can be challenging and most cytopathologists outside of major academic centers may see fewer than one or two cases a decade. The clinician should be alert to the possibility that MTC is incorrectly labeled as a parathyroid tumor, poorly differentiated carcinoma of
unknown etiology, or rarely anaplastic carcinoma. It is important for the clinician, who may have a broader perspective, to request stains for CT, CEA, or chromogranin A to cue the cytopathologist to the correct diagnosis.
unknown etiology, or rarely anaplastic carcinoma. It is important for the clinician, who may have a broader perspective, to request stains for CT, CEA, or chromogranin A to cue the cytopathologist to the correct diagnosis.
Metastasis to local lymph nodes occurs frequently. Eighty percent of patients with a palpable MTC (>1 cm) have lymph node metastasis (76,77,78). Furthermore, lymph node metastasis is frequently not apparent to the operating surgeon and may be overlooked by the pathologist unless each node removed is carefully examined. The most common pattern of lymph node metastasis is to ipsilateral nodes in levels II-VI; metastasis to contralateral nodes occurs in approximately 40% of cases where the primary MTC is palpable (76,77). A common pattern of metastasis is to mediastinal lymph nodes. Hepatic and pulmonary parenchymal metastases are commonly vascular. Clinicians should be aware that even experienced radiologists will commonly label a small focus of hepatic metastasis, defined by contrast-enhanced CT scanning, as a hemangioma. Another common pattern of metastasis is military spread of MTC to the liver. This pattern of metastasis is difficult to identify by any currently available imaging technique, but may be identified by laparoscopic evaluation of the liver (79) Bone metastasis are most commonly lytic; spinal metastasis are not uncommon and of particular concern because of the potential for spinal cord involvement.
Hereditary MTC
The clinical syndromes associated with MTC have been discovered and rediscovered over the past century. However, it was Sipple’s description in 1961 that provided the first compilation of the association between thyroid cancer and pheochromocytoma (now known as MEN2A) (2,80). The subsequent separation of MEN1 from MEN2 (81) and the separation of MEN2A and MEN2B (82,83,84,85,86) further defined the boundaries between these clinical syndromes. The addition of several variants: Familial MTC (without other manifestations of MEN2A) (87), MEN2A with Hirschsprung disease (88), and MEN2A with cutaneous lichen amyloidosis (71), (70) has added further complexity to the classification system. The discovery of RET mutations and insight gained from the correlation of clinical phenotype with specific molecular changes has led to the current classification system (Table 1) (89).
Table 51.1 Hereditary Medullary Thyroid Carcinoma | |
|---|---|
|
Multiple Endocrine Neoplasia type 2A
The association of MTC, pheochromocytoma, and hyperparathyroidism is classified as MEN2A (Table 51.1). The fully formed clinical syndrome, described by Sipple, develops most commonly in the third and fourth decades. It is characterized by bilateral and multicentric MTC in more than 90% of gene carriers, unilateral or bilateral multicentric pheochromocytomas in one-half and hyperparathyroidism in 5% to 10%. Prior to the broad recognition of this syndrome, death was as likely to occur from sudden death caused by a pheochromocytoma as from metastatic thyroid carcinoma. Familiarity with the clinical syndrome and its genetic nature has led to earlier recognition of this disorder in affected families. It is characteristically transmitted as an autosomal dominant trait. The fact that both MTC and pheochromocytoma manifest late in the second or the third or fourth decade insured transmittal of this gene to the next generation and the existence of multigenerational kindreds; death from either MTC or pheochromocytoma most commonly occurred in the mid 50s, although earlier death does occur. As the average life expectancy at the beginning of the 20th century was 50 to 55 years, it is not surprising that the hereditary nature of this syndrome escaped detection.
Following or concurrent with Sipple’s description in 1961 there followed in rapid-fire sequence the discovery of CT (6,8), the recognition that the parafollicular thyroid cells produce CT, the recognition that MTC is composed of transformed C cells (90), and the recognition that calcium and other small peptides such as glucagon (91) and gastrin (50,92,93) (or pentagastrin) stimulate release of CT from the C cell. These observations led to the use of calcium and/or pentagastrin to diagnose early MTC. More importantly, early detection provided the rationale and method for detection of early C cell abnormalities and the definition of the spectrum of C cell abnormalities found in MTC.
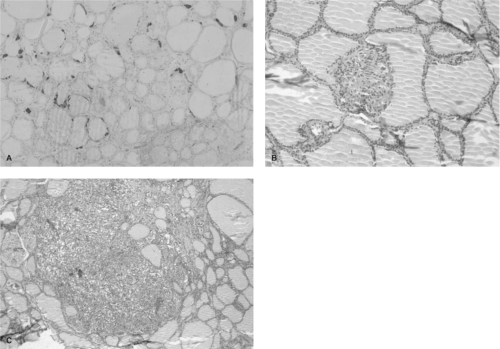 Figure 51.4 Progression of transformation in hereditary MTC. Panel A shows a histologic section from 3-year-old child with a codon 634 mutation. The C cells are stained for calcitonin using immunohistochemical techniques. The section shows a slight increase of C cell number, clustering around the thyroid follicles. Panel B shows “nodular” C cell hyperplasia. In this histologic section the C cells displace the thyroid follicle, creating the appearance of a “nodule.” Panel C shows a focus of microscopic medullary thyroid carcinoma characterized by obliteration of the thyroid follicular structure and coalescence of several foci of hyperplastic C cells. See color plate. |
The Natural History of MTC in MEN2A
The earliest histologic abnormality in the thyroid glands removed from children and young adults with MEN2A is single or multicentric C cell hyperplasia (Fig. 51.4) (94). These individual foci evolve through a predictable histologic state described as “nodular” hyperplasia (so named because the growing mass of C cells appears to displace a thyroid follicle (68), creating the appearance of a nodule), to microscopic MTC, and then a visible focus of MTC (68). Although the progression time is unclear, foci of microscopic MTC have been observed in children as young as 10 (95) and 12 months of age; however, there is considerable variability of the latency period for the development of cancer (up to 50 years) in the majority of MEN2A cases. This variability suggests that the presence of a mutant RET tyrosine kinase receptor is necessary but not sufficient for transformation.
It is unclear at exactly what point metastasis occurs in this evolution; the youngest identifiable published example of metastatic disease to local lymph nodes is in a 6-year-old child, (96,97) however there are unpublished examples as young as 3 years of age in the context of MEN2A. It is also unclear at what point metastasis progresses beyond local lymph nodes. There are reports dating from the early 1970s that patient with hereditary MTC and local lymph node metastasis may be cured by extensive lymph node dissection. The authors’ personal long-term experience with a number of patients with MTC and lymph node metastasis without evidence of metastatic disease 5 to 30 years following surgery (78,98,99) provide a compelling argument that patients with local metastatic disease can be cured by lymph node dissection (100,101,102).
Pheochromocytoma in MEN2A
Approximately one-half of individuals with MEN2A will develop clinical evidence of a pheochromocytoma. These tumors are multicentric and evolve out of a background of adrenal medullary hyperplasia. A detailed discussion of pheochromocytoma in MEN2 is beyond the scope of this chapter and the reader is referred to other reviews (103,104,105,106). Several points are of relevance to the management of MTC in this context. First, all patients with MEN2A should be screened for pheochromocytoma prior to consideration of thyroid surgery. Six to 8% of patients with apparently sporadic MTC will have hereditary MTC (15). Measurement of plasma or urinary metanephrines and catecholamines is sufficient for screening purposes. More commonly the question of pheochromocytoma is raised in the context of a planned thyroid surgical procedure where delay is opposed by the patient and/or surgeon. In this situation, demonstration of normal adrenal glands by a high quality CT scan will suffice as 99% of MEN2A pheochromocytomas are intra-adrenal or within the abdomen. If abnormal metanephrines or catecholamines or radiographic abnormalities of either adrenal gland are identified, the thyroid surgical procedure should be deferred until the pheochromocytoma is removed or the etiology of the imaging abnormality is clarified. A second point is that the absence of hypertension does not exclude MEN2-related pheochromocytomas. Unlike the clinical picture in sporadic pheochromocytoma or other hereditary pheochromocytoma syndromes (von Hippel–Lindau disease or hereditary paraganglioma), the disproportionate production of epinephrine associated with MEN2-related pheochromocytomas is more likely to produce palpitations than hypertension (Fig. 51.5).
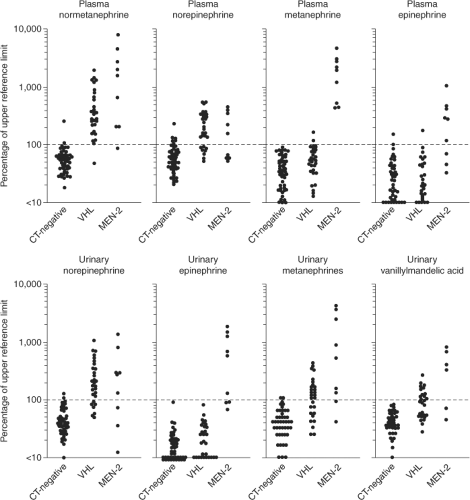 Figure 51.5 Plasma Concentrations of Normetanephrine, Norepinephrine, Metanephrine, and Epinephrine (Upper Panels) and Urinary Excretion of Norepinephrine, Epinephrine, Metanephrines, and Vanillylmandelic Acid (Lower Panels). The values are expressed as percentages of the upper reference limit for each test. Data on individual patients are shown for three groups of patients with von Hippel–Lindau disease and multiple endocrine neoplasia type 2 (MEN-2), as follows: Patients with von Hippel–Lindau disease or MEN-2 in whom a pheochromocytoma was ruled out on the basis of normal computed tomography (CT-negative), patients with von Hippel–Lindau disease who had histologically verified pheochromocytomas (VHL), and patients with MEN-2 who had histologically verified pheochromocytomas (MEN-2). The values for patients with pheochromocytoma were determined when the tumors were first identified by computed tomography. The dotted horizontal line represents the upper reference limit for each test. The scales are logarithmic. (Data from Eisenhofer G, Lenders JWM, Linehan WM, et al. Plasma normetanephrine and metanephrine for detecting pheochromocytoma in von Hippel–Lindau disease and multiple endocrine neoplasia type 2. Reprinted with permission from N Engl J Med 340: 1872–1879, 1999. |
Hyperparathyroidism in MEN2A
Hyperparathyroidism is an uncommon manifestation of MEN2A (5% to 10%). It is appropriate to measure a serum
calcium concentration preoperatively in patients with MTC and the possibility of MEN2 should be considered in those with an elevated value.
calcium concentration preoperatively in patients with MTC and the possibility of MEN2 should be considered in those with an elevated value.
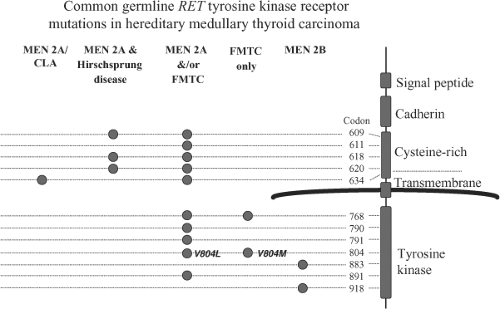 Figure 51.6 Common germline RET proto-oncogene tyrosine kinase receptor mutations in hereditary medullary thyroid carcinoma. The figure shows genotype-phenotype correlation for the most common mutations found in multiple endocrine neoplasia type 2. Each of specific mutations is described in Table 51.2. MEN2A/CLA, multiple endocrine neoplasia type 2A and cutaneous lichen amyloidosis; MEN2A/Hirschsprung disease, the association of MEN2A with Hirschsprung disease in affected kindreds; FMTC, familial medullary thyroid carcinoma without other manifestations of MEN2A; MEN2B, multiple endocrine neoplasia type 2B. |
Variants of MEN2A
In addition to classic MEN2A, there are 3 distinct variants of MEN2A. The first, familial MTC (FMTC) (87), is characterized by the presence of MTC without other manifestations of MEN2A. The reports describing this variant have generally encompassed three or four generations with 20 to 30 affected members with no evidence of pheochromocytoma or hyperparathyroidism. MTC in the context of FMTC tends to develop later and be of lower aggressiveness. Before categorizing a particular kindred with the diagnosis of FMTC, it is important to consider the fact that the penetrance of pheochromocytoma in MEN2A is only 50%. This can lead to miscategorization of
a small kindred with predominantly young members and no history of pheochromocytoma as FMTC. The important point is that screening for pheochromocytoma should continue in affected members unless there is a multigenerational experience to exclude this possibility.
a small kindred with predominantly young members and no history of pheochromocytoma as FMTC. The important point is that screening for pheochromocytoma should continue in affected members unless there is a multigenerational experience to exclude this possibility.
Multiple endocrine neoplasia type 2 with Hirschsprung disease is also an uncommon variant in which affected family members develop Hirschsprung disease (88,107) The penetrance varies considerably; in some kindreds a high percentage are affected; in other equally large kindreds only one or two individuals may be affected (15,108).
The association of a cutaneous form of lichen amyloidosis with MEN2A is the most recently discovered variant (71,70). In a small number of kindreds, estimated to be fewer than 50 world-wide, a pruritic cutaneous lesion develops invariably over the upper back. The lesion may be unilateral or bilateral and may extend from C6 to approximately T5 vertebral levels. Current evidence suggests that it is the continuous scratching and irritation of the pruritic region, a condition analogous to “friction amyloidosis,” (109) that causes the cutaneous skin lesion (110). In some kindreds, the presence of localized pruritus is a phenotypic indicator of MEN2A (70).
Multiple Endocrine Neoplasia Type 2B
The association of MTC, pheochromocytoma, and ganglioneuromatosis has been classified as MEN2B. Williams and Pollock (111) were the first to piece together the components of this syndrome, although earlier reports exist. It is transmitted as an autosomal dominant trait (83,112,113), although most identified cases represent de novo mutations (114). The presence of mucosal neuromas on the distal tongue, subconjunctiva, and throughout the gastrointestinal tract give this clinical syndrome its most distinctive feature. Since the mucosal neuromas on the distal tongue and subconjunctiva appear early in life, the diagnosis of MEN2B may be suspected during infancy. Hyperparathyroidism is almost never found (115), but unilateral or bilateral pheochromocytomas, analogous to those found MEN2A occur in approximately one-half of affected patients.
This clinical syndrome is most commonly identified in childhood as a result of gastrointestinal manifestations that may include colic, abdominal cramping, intestinal obstruction (pseudo or real), or diarrhea. These symptoms are most commonly related to intestinal dysfunction caused by neurologic abnormalities of the GI tract (Hirschsprung-like) or intermittent physical obstruction caused by neuromas (85). Marfan-like features in these patients include long, thin arms and legs, a reduced upper/lower body ratio, pectus excavatum, and slipped femoral capital epiphyses (83) The lens and aortic manifestations of Marfan syndrome are never seen. Approximately 5% will have a more subtle form of the disorder with normal stature or barely detectable neuromas making clinical diagnosis challenging.
MTC in MEN2B is bilateral and multicentric, metastasizes early and is generally more aggressive than that found in MEN2A (116,117). Metastasis to local lymph nodes during the first few years of life is common. Despite the generally high level of aggressiveness, there is considerable variability in the clinical outcome. Death from metastatic MTC most commonly occurs during the second or third decade, but there are well-described examples of patients with metastatic disease who have had prolonged survival (113,118).
Table 51.2 Mutations of the Ret Proto-Oncogene in Hereditary Medullary Thyroid Carcinoma | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


