MEDULLARY THYROID CARCINOMA
Part of “CHAPTER 40 – THYROID CANCER“
Medullary thyroid carcinoma (MTC), first recognized in 1959, is a pleomorphic neoplasm with amyloid struma that arises from the calcitonin-secreting C cells of the thyroid. Approximately 20% are familial tumors that are transmitted as an autosomal dominant trait and are often associated with other endocrine neoplasms. The genes responsible for the familial forms of MTC map to the pericentromeric region of chromosome 10.186
MULTIPLE ENDOCRINE NEOPLASIA TYPE 2 SYNDROMES
MTC may occur in four settings. Sporadic MTC is a unilateral thyroid tumor with no somatic lesions other than the neoplasm. Multiple endocrine neoplasia type 2A (MEN2A) is familial, bilateral MTC with hyperparathyroidism and bilateral pheochromocytoma. Multiple endocrine neoplasia type 2B (MEN2B) may be sporadic or familial and features bilateral MTC, bilateral pheochromocytoma, an abnormal phenotype with multiple mucosal ganglioneuromas, and musculoskeletal abnormalities suggestive of the marfanoid habitus. Familial non-MEN MTC (FMTC) comprises bilateral MTC with no other endocrine tumors or somatic abnormalities.187 MEN2A is always inherited as an autosomal dominant trait, whereas MEN2B may be similarly transmitted or can occur sporadically. FMTC is an autosomal dominant trait that is the least common form, and manifests at a later age.187
Medullary Thyroid Carcinoma.
The MTC in both MEN2 syndromes is generally bilateral and multicentric, as opposed to sporadic MTC, which is usually unilateral. The C cells are normally located in the upper and middle thirds of the lateral thyroid lobes. In MEN2, the C cells initially undergo hyperplasia, which precedes the development of MTC.
Parathyroid Disease.
One-third to one-half of the patients with the MEN2A syndrome have hyperparathyroidism, most of whom (85%) have parathyroid hyperplasia.188 In contrast, almost no patients with MEN2B have parathyroid disease.189 Patients with MEN2A seldom present with symptoms of hypercalcemia but often form kidney stones. Parathyroid hyperplasia is discovered during MTC surgery in most MEN2A patients, even those without clinical or biochemical evidence of hyperparathyroidism.189
Pheochromocytoma.
Adrenal medullary disease, ranging from diffuse or nodular hyperplasia to large bilateral multilobular pheochromocytomas, occurs in both MEN2 syndromes. Pheochromocytomas or medullary hyperplasia is typically bilateral and occurs in ˜40% of patients, with a range of 6% to 100% in different kindreds.188 The symptoms are typically subtler than those encountered with sporadic pheochromocytoma.190 The diagnosis is usually established by demonstrating high urinary or plasma catecholamine levels, but the total urinary catecholamines may be normal, with only an increased epinephrine:norepinephrine ratio, particularly in those with adrenal medullary hyperplasia190 (see Chap. 86).
Multiple Endocrine Neoplasia Type 2B.
MEN2B is characterized by a constellation of somatic abnormalities consisting of ganglioneuromas of the tarsal plates and the anterior third of the tongue, and a marfanoid habitus. Nodules may occur in the lips, causing them to appear lumpy and patulous. Ganglio-neuromas in the alimentary tract may be associated with constipation, diarrhea, and megacolon. The marfanoid characteristics include long limbs, hyperextensible joints, scoliosis, and anterior chest deformities, but not ectopic lens or cardiovascular abnormalities189 (see Chap. 188).
Point mutations of the RET protooncogene have been identified in germline and tumor DNA of unrelated patients from kindreds with MEN2A, MEN2B, and FMTC.186,191,192 A relationship exists between specific RET protooncogene mutations and the disease phenotype in MEN2.192 Although several different and independent point mutations in the genomic sequence of the RET protooncogene have been identified, all involving codons for cysteine residues, the normal function of RET is not yet known, and its role in the development of these inherited neoplasms remains unclear. Nevertheless, the identification of point mutations provides an important direct means of identifying affected MEN and FMTC kindreds.
PREVALENCE
MTC accounts for ˜10% of all thyroid malignancies. Approximately 80% to 90% of MTC cases occur sporadically, and 10% to 20% are inherited. MTC may occur at any age, but sporadic disease is diagnosed later in life than is familial MTC. The median age of patients seen at the Mayo Clinic with sporadic MTC was 51 years, compared with 21 years for those with familial tumors.190 MEN2A and FMTC are both characterized by the development of bilateral MTC, but the age at onset of FMTC is usually later, ranging from 40 to 50 years, as compared with an average of 20 to 30 years for MEN2A. Familial MTC occurs with equal frequency in both sexes, while sporadic MTC has a female-male ratio of 1.5:1.
PATHOLOGY
Sporadic and familial MTC are histologically similar, although a wider spectrum of appearances is encountered in familial tumors, ranging from isolated hypertrophied C cells to large bilateral multicentric tumors that are usually in the superior portions of the thyroid lobes. Typically, the tumor is composed of fusiform or polygonal cells surrounded by irregular masses of amyloid and abundant collagen (Fig. 40-11). Calcifications
are present in approximately one-half of the tumors, and occasionally trabecular bone formation is seen. Calcitonin can usually be demonstrated in the tumor by immunohistochemical studies.
are present in approximately one-half of the tumors, and occasionally trabecular bone formation is seen. Calcitonin can usually be demonstrated in the tumor by immunohistochemical studies.
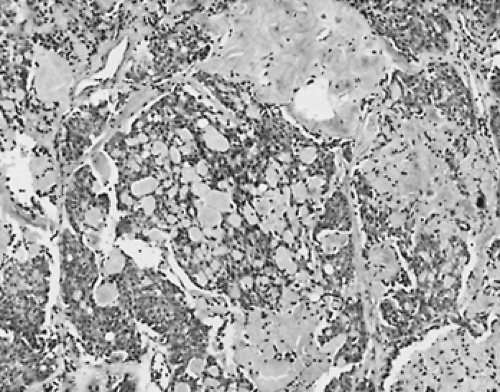 FIGURE 40-11. Medullary thyroid carcinoma showing trabecular architecture with spindle and round cells and abundant stroma that stained positively with Congo red, for amyloid. ×100 |
Cervical node metastases occur early in the disease and can be seen with primary lesions as small as several millimeters. Tumors >1.5 cm in diameter are more likely to metastasize to distant sites.
The pattern of tissue calcitonin staining may differentiate virulent from less aggressive tumors. In one study, patients with primary tumors that showed intense homogeneous calcitonin staining were all clinically well on follow-up examination, whereas patients whose tumors showed patchy localization of calcitonin either developed metastatic disease or died of cancer within 6 months to 5 years of initial surgery.193
DIAGNOSIS
Clinical Features.
Patients with sporadic disease or previously unrecognized familial MTC usually present with one or more painless thyroid nodules in an otherwise normal gland, but the tumor may cause pain, dysphagia, and hoarseness. The dominant sign may be enlarged cervical lymph nodes or, occasionally, distant metastases, most commonly to the lung, followed in frequency by metastases to the liver, bone (osteolytic or osteoblastic lesions), and brain. In approximately one-half of patients with sporadic MTC, cervical lymph node metastases are present at the time of diagnosis. The thyroid nodule may be cold or normal on radionuclide imaging and is usually solid on echography and malignant on FNA. Radiographs may show dense, irregular calcifications of the primary tumor and cervical nodes, and mediastinal widening that is due to metastases. Rarely, an abnormal phenotype may correctly identify the tumor, or a paraneoplastic syndrome may be the presenting manifestation.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


