MTC risk group
Exon
Codon
Recommended age for genetic testing
Highest risk
16
918
As soon as possible (first year of life)
High-risk
11
634
3 years
15
883
3 years
Moderate risk
8, 13–15
533, 609, 611, 618, 620, 790, 804, 891
5 years
Pheochromocytoma risk (%) | Exon | Codon | Recommended age for biochemical screening (years) |
|---|---|---|---|
~50 | 16, 11 | 918, 833, 634 | 11 |
~20 | 10 | 609, 611, 618, 620 | 16 |
~5 | 13–15 | 804, 891 | 16 |
PHPT risk | Exon | Codon | Recommended age for biochemical screening (years) |
|---|---|---|---|
~20 % | 11 | 634 | 11 |
~5 % | 10, 13–15 | 609, 611, 618, 620, 804, 891 | 16 |
No | 16 | 918 | – |
4.1 Highest Risk Mutation (RET M918T)
Patients with codon M918T mutation and MEN2B have the highest risk of aggressive MTC occurring at a young age. Microscopic MTC was reported in a RET M918T gene carrier at 9 weeks of age (Shankar et al. 2012), and lymph node metastases have been diagnosed as early as 3 months of age (Zenaty et al. 2009). The vast majority (90–95 %) of patients carrying RET 918 mutations have de novo mutations (Brauckhoff et al. 2014) of paternal origin (Carlson et al. 1994). Because of this high proportion of de novo M918T mutations, early diagnosis via genetic screening is rarely possible. Similar to findings in the large series of Brauckhoff et al. (2014), in our own series of 21 patients with RET 918 mutations, the median age at MTC diagnosis, 13.9 years (0.9–30 years), was far too late. In both series, only about 20 % of patients were cured.
There are insufficient data concerning the clinical behavior of MTC in patients who have RET A883F mutations or in patients with double mutations involving RET codon V804M and either codon Y806C, S904C, E805 K, or Q781R.
4.2 High-Risk Group (RET 634; RET 883)
Mutations in RET codon 634 are characterized by an early age of MTC onset, and there is good evidence that there is significant age-related progression from CCH to MTC (Machens et al. 2003). Microscopic MTC has been detected in RET 634 mutation carriers as early as 10 months of age (Zenaty et al. 2009), and a larger series demonstrated that MTC associated with any mutation at codon 634 commonly appears before 10 years of age but is rarely associated with lymph node metastases in patients younger than 14 years (Machens et al. 2003).
4.3 Moderate Risk Group (Exon 10 RET 609, 611, 618, 620, and Exons 13–15)
The clinical course of MTC in patients with mutations in codons other than codons 634/918 varies widely. With lower risk RET mutations, the lifetime MTC risk is high, typically shows later onset, and is less aggressive compared with the high-risk and highest risk groups (Rich et al. 2014). The earliest ages at which MTC was reported in patients with RET 609, 611, 618, and 620 mutations (identified by family screening) were 5, 15, 8, and 5 years, respectively, while lymph node metastases were reported at the ages of 39, -, 11, and 10, respectively (Rich et al. 2014). For exon 10 mutations, the median age at manifestation of MTC is between 20 and 40 years, and lymph node metastasis rarely occurs before 30 years of age (Frank-Raue et al. 2011). The results of a multicenter study of 340 patients carrying exon 10 mutations show differences in the aggressiveness of MTC depending on the mutated codon (Frank-Raue et al. 2011). In order of severity from most to least severe, mutations in codons 620, 618, 611, and 609 result in earlier age at manifestation, more advanced tumor stage at manifestation, and decreased chance of cure. In carriers of RET mutations 533 and 804 that were identified by family screening, the earliest manifestations of MTC were reported at 21 and 16 years of age, respectively, and the median ages were 42 and 52 years, respectively. For carriers of RET mutations 533 and 804, the earliest ages at lymph node metastases were 22 and 31 years, with median ages of 59 and 53 years (Rich et al. 2014). The cumulative risk of MTC in children at the age of 20 was 10 % or lower for mutations in codons 533, 609, 611, 791, and 804 (Rich et al. 2014).
RET mutations Y791F and S649L are described as having variable phenotypes that are in most cases very mild (Berndt et al. 1998; Gimm et al. 2002; Frank-Raue et al. 2008). Accordingly, some questioned the pathogenicity of these RET mutations (Erlic et al. 2010; Toledo et al. 2015). In these mutations, thyroidectomy should not be based on mutation status alone but also on elevated CTN levels.
5 Clinical Implications
5.1 RET Testing
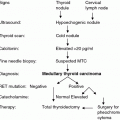
The benefits of RET testing are so well described that this genetic test is considered the standard of care in all patients with newly detected MTC and in all first-degree relatives of a patient with a known RET mutation. A family history is often inadequate for establishing the diagnosis of familial disease, and a more thorough evaluation by genetic and biochemical screening often reveals a family history of MTC in patients originally thought to have the sporadic form of the disease. About 1–7 % of apparently sporadic cases have identifiable RET mutations, including about 2–9 % that have de novo germline mutations (Kloos et al 2009). Presymptomatic identification of a RET mutation carrier is the predominant route to an MEN2 diagnosis in children. The most common practice for RET analysis includes DNA sequencing of exons 5, 8, 10, 11, and 13–16. It makes sense to perform targeted analysis of exons 15 and 16 in patients with the MEN2B phenotype, but analysis of the entire coding sequence of the RET gene should be reserved for patients with strong evidence suggesting a hereditary cause of the disease and negative findings when sequencing exons 5, 8, 10, 11, and 13–16. Prior to genetic testing, an experienced clinician or genetic counselor should communicate the risks, benefits, and limitations of this test. Thereafter, the patient or the patient’s parents can provide informed consent. In families with hereditary MTC, genetic testing should be performed as soon as possible after birth for the highest risk group of RET mutations, at 3 years of age or before in children in the high-risk category, and at 5 years of age in children in the moderate risk category.
During recent years, there has been a change in the spectrum of RET mutations detected in patients with hereditary MTC. Specifically, there has been a shift from the “classical” mutation at codon 634 in exon 11 to more cases with mutations in exons 13–15 and less aggressive disease (Frank-Raue et al. 2007). Initially, the frequency of diagnosed RET mutations in patients with MEN2A was 85 % mutations in codon 634, with mutations in codons 609, 611, 618, and 620 accounting for the additional 10–15 % of cases (Eng et al. 1996). Our recent analysis of the RET proto-oncogene in patients with hereditary MTC provides evidence for this change in the spectrum of detected mutations. Exon 13–15 mutations, so-called rare mutations, were diagnosed in 39 % of families, exon 10 and 11 mutations in 54 %, and exon 16 mutations in 6 %. This change in the frequency of diagnosed mutations in MEN2A families from high-risk mutations to the so-called rare mutations in codons 13, 14, and 15 may impact the overall prognosis of hereditary MTC, i.e., improve the overall prognosis of hereditary MTC. The reasons underlying this change in mutation detection may include the routine RET diagnostics in all patients diagnosed with MTC, the discovery of hereditary cases in apparently sporadic (4–7 %) cases, and the extension of the analyses to included mutations other than the known “hot spots” (Berndt et al. 1998). In addition, there is a distinct distribution of RET mutations in different parts of the world that depend on the genetic background of the local population, which may also affect the detection rates of specific RET mutations.
5.2 Prophylactic Thyroidectomy
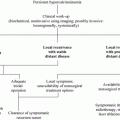
Prophylactic or early thyroidectomy is the only curative therapy for MTC in RET gene carriers. The biological behavior of MTC, meaning the age at onset of MTC plus the aggressiveness of the disease, is mutation-dependent. These findings have led to stratification of RET mutations according to risk level (i.e., highest, high, and moderate), but heterogeneity in disease expression and progression within these groups varies considerably, especially in the moderate risk group. The optimal timing for thyroidectomy is a major consideration: Should the timing for a given RET mutation be based on the typical behavior of MTC in carriers of this RET mutation or on the earliest reported age at which MTC/metastases occur? Is the aim true prophylactic thyroidectomy (before manifestation of MTC) or is the aim long-term “biochemical” cure? Current discussions about performing prophylactic thyroidectomy in the first few years of life include: (i) concerns that the surgical and anesthetic risks outweigh the benefits of surgery at that age; (ii) concern for iatrogenic hypothyroidism (Frank-Raue et al. 2006); and (iii) the effectiveness of CTN for detecting clinically relevant MTC.
5.3 The Effectiveness of CTN for Detecting Clinically Relevant MTC
If the aim of prophylactic thyroidectomy is to remove the thyroid before MTC metastases occur and, concurrently, to decrease potential medical and surgical morbidity, the effectiveness of detecting clinically relevant MTC by CTN determination becomes crucial. Given our evolving understanding of MEN2A-related MTC, the decision regarding the age of prophylactic thyroidectomy is no longer based upon genotype alone. Rather, this decision is currently based on additional clinical data, the most important being basal or stimulated serum CTN levels. Data from the current literature show that basal CTN levels below 40 pg/ml and below 60 pg/ml are not associated with lymph node metastases; specifically, studies from France, Germany, and Italy show that all patients were disease-free after surgery when the basal pre-operative serum CTN level was below 40 pg/ml (Elisei et al. 2012; Machens et al. 2009; Rohmer et al. 2011).
Therefore, current guidelines state that the timing of prophylactic thyroidectomy should be based on the risk level of the particular RET mutation and on the CTN level; specifically, the decision to perform thyroidectomy should err on the safe side if the CTN level is elevated but below 30 pg/ml, especially in the moderate risk group. The recent version of the American Thyroid Association (ATA) guidelines included three risk groups for RET mutations, i.e., highest risk, high-risk, and moderate risk categories. Children in the ATA highest risk category should undergo thyroidectomy in their first year of life, and perhaps even in their first months of life. Children in the ATA high-risk category should have ultrasound of the neck and CTN measurement performed prior to thyroidectomy. Thyroidectomy should typically be performed at 5 years of age or earlier, depending on the presence of elevated serum CTN levels. Children in the ATA-moderate category should have ultrasound of the neck and measurement of CTN prior to thyroidectomy. Thyroidectomy timing should most often be based on serum CTN levels; however, annual or biannual evaluations may extend the timing by several years or even decades (Wells et al. 2015).
5.4 Screening for Pheochromocytoma or PHPT
In addition to early treatment of MTC, the main concerns in a RET gene carrier are not to miss a pheochromocytoma diagnosis, because often there is a risk of lethal pheochromocytoma crisis during neck surgery or during childbirth, and to avoid overlooking primary HPT before neck surgery. Screening for pheochromocytoma should begin at the age of 11 for children carrying RET mutations in exons 16, 15, and 11 in codons 918, 833, and 634 and by 16 years of age in children carrying RET mutations in exons 5, 8, 10, and 13–15, e.g., in codons 609, 611, 618, 620, 804, and 891 (Table 1). Screening consists of measuring plasma levels of free metanephrines and normetanephrines, or 24-hour urine metanephrines and normetanephrines. Adrenal imaging with computerized tomography or MRI is indicated in patients with positive biochemical results. For practical reasons, patients should be screened for HPTH at the time of screening for pheochromocytoma; in MEN2B, no HPTH screening is necessary (Wells et al. 2015).
References
Barbet J, Campion L, Kraeber-Bodere F, Chatal JF, Group GTES (2005) Prognostic impact of serum calcitonin and carcinoembryonic antigen doubling-times in patients with medullary thyroid carcinoma. J Clin Endocrinol Metab 90(11):6077–6084CrossRefPubMed
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


