Benign proliferation of the glandular tissue of the male breast constitutes the histologic hallmark of gynecomastia, which, if sufficiently great, appears clinically as palpable or visual enlargement of the breast. This condition, which is exceedingly common, may (a) be a sign of a serious underlying pathologic condition, (b) cause physical or emotional discomfort, or (c) be confused with other breast problems, most significantly carcinoma.
PREVALENCE
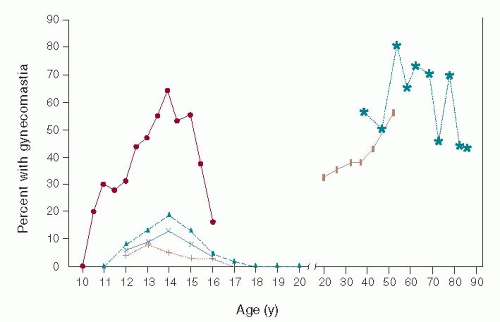
Breast glandular proliferation commonly occurs in infancy, during puberty, and in older age. It has been estimated that between 60% and 90% of infants exhibit the transient development of palpable breast tissue owing to estrogenic stimulation from the maternal-placental-fetal unit. This stimulus for breast growth ceases as the estrogens are cleared from the neonatal circulation, and the breast tissue gradually regresses over a 2- to 3-week period, but may persist longer. Although population studies have shown that the prevalence of pubertal gynecomastia varies widely, most have indicated that 30% to 60% of pubertal boys exhibit gynecomastia, which usually begins between 10 and 12 years of age, with the highest prevalence between 13 and 14 years of age (corresponding to Tanner stage III or IV of pubertal development), followed by involution that is usually complete by age 16 to 17 years (1). The percentage of men who exhibit gynecomastia increases with advancing age, with the highest prevalence found in the 50- to 80-year age range (Fig. 8-1). The prevalence of the condition in men ranges between 24% and 65%, with the differences between series being accounted for by the defining criteria and by the population studied (2).
PATHOGENESIS
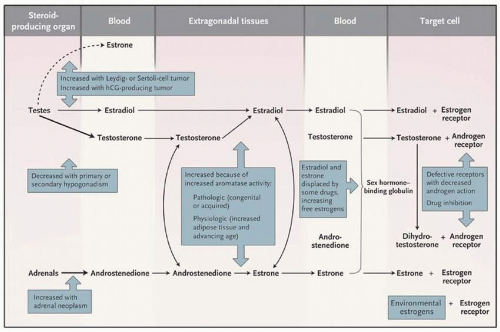
No inherent differences appear to exist in the hormonal responsiveness of the male or female breast glandular tissue (3). The hormonal milieu, the duration and intensity of stimulation, and the individual’s breast tissue sensitivity determine the type and degree of glandular proliferation. Under the influence of estrogens, the ducts elongate and branch, the ductal epithelium becomes hyperplastic, the periductal fibroblasts proliferate, and the vascularity increases. This histologic picture is found early in the course of gynecomastia and is often referred to as the florid stage. Acinar development is not seen in the male because it requires the presence of progesterone in concentrations found during the luteal phase of the menstrual cycle (3). Androgens exert an antiestrogen effect on rodent breast cancer models and the human MCF-7 breast cancer cell line; they are thought to antagonize at least some of the effects of estrogens in normal breast tissue (4). Accordingly, gynecomastia is usually considered to represent an imbalance between the breaststimulatory effects of estrogen and the inhibitory effects of androgens. In fact, alterations in the estrogen-to-androgen ratio have been found in many of the conditions associated with gynecomastia. Such alterations can occur through a variety of mechanisms (Table 8-1; Fig. 8-2).
In men, the testes secrete 95% of the testosterone, 15% of the estradiol, and less than 5% of the estrone produced daily. Most of the circulating estrogens are derived from the extraglandular conversion of estrogen precursors by extragonadal tissues, including the liver, skin, fat, muscle, bone, and kidney (Fig. 8-2). These tissues contain the aromatase enzyme that converts testosterone to estradiol and androstenedione, an androgen primarily secreted by the adrenal glands, to estrone. Estradiol and estrone are interconverted in extragonadal tissues through the activity of the 17-ketosteroid reductase enzyme. This enzyme is also responsible for the interconversion of testosterone and androstenedione. When androgens and estrogens enter the circulation, either through direct secretion from gonadal tissues or from the sites of extragonadal metabolism, most are bound to sex hormone-binding globulin (SHBG), a protein derived primarily from the liver and one that has a greater affinity for androgens than for estrogens. The non-SHBG sex hormones circulate either in the free or unbound state or are weakly bound to albumin. These fractions are able to cross the plasma membrane of target cells and are bound to steroid receptors. Testosterone and dihydrotestosterone bind to the same hormone-responsive element. Each also binds to the hormone-responsive element of the appropriate genes, resulting in the initiation of transcription and hormone action. A similar sequence of events occurs after the binding of estradiol or estrone to the estrogen receptor (5).
FIGURE 8-1Prevalence of gynecomastia at various chronologic ages. Data were derived from multiple population studies. (Adapted from Braunstein GD. Pubertal gynecomastia. In: Lifshitz F, ed. Pediatric endocrinology. New York: Marcel Dekker, 1996:197-205, with permission.)
TABLE 8-1 Conditions Associated with Gynecomastia and Their Primary Pathophysiologic Mechanisms
Increased aromatization (peripheral and glandular)
Sertoli cell (sex cord) tumors
Testicular germ cell tumors
Leydig cell tumors
Adrenocortical carcinoma
Hermaphroditism
Obesity
Hyperthyroidism
Liver disease
Testicular feminization
Refeeding after starvation
Primary aromatase excess
Displacement of estrogen from sex hormone-binding globulin
Spironolactone
Ketoconazole
Decreased estrogen metabolism
Cirrhosis
Exogenous sources
Topical estrogen creams and lotions
Ingestion of estrogen
Tree tea or lavender oils
Eutopic hCG production
Choriocarcinoma
Ectopic hCG production
Lung carcinoma
Liver carcinoma
Gastric carcinoma
Kidney carcinoma
Decreased testosterone synthesis
Primary gonadal failure, congenital
Anorchia
Klinefelter’s syndrome
Hermaphroditism
Hereditary defects in testosterone synthesis
Primary gonadal failure, acquired
Viral orchitis
Castration
Granulomatous diseases (including leprosy)
Testicular failure owing to hypothalamic or pituitary disease
Androgen resistance owing to androgen receptor defects
Other
Chronic renal failure
Chronic illness
Spinal cord injury
Human immunodeficiency virus
Enhanced breast tissue sensitivity
hCG, human chorionic gonadotropin.
Adapted with permission from Mathur R, Braunstein GD. Gynecomastia: pathomechanisms and treatment strategies. Horm Res 1997;48:95-102.
FIGURE 8-2 Pathways of estrogen and androgen production, action, and metabolism, and pathologic and physiologic changes that alter the pathways. (Adapted from Braunstein GD. Gynecomastia. N Engl J Med 2007;357:1229-1237, with permission.)
From a pathophysiologic standpoint, an imbalance between estrogen and androgen concentrations or effects can occur as a result of abnormalities at several levels (Table 8-1; Fig. 8-2). Overproduction of estrogens from testicular or adrenal neoplasms or enhanced extraglandular conversion of estrogen precursors to estrogens can elevate the total estrogen concentration. Such extraglandular conversion can occur directly in the breast tissue. Indeed, increased aromatization of androgens to estrogens has been noted in pubic skin fibroblasts from some patients with idiopathic gynecomastia (6). Elevations of the absolute quantity of circulating free estrogens can occur if estrogen metabolism is slowed or if SHBG-bound estrogens are displaced from the protein. Conversely, decreased secretion of androgens from the testes—caused primarily by defects in the testes or secondary to loss of tonic stimulation by pituitary gonadotropins, enhanced metabolic degradation of androgens, or increased binding of androgens to SHBG—results in decreases in free androgens that could antagonize the effect of estrogens on the breast glandular tissue. As noted previously, androgen and estrogen balance depends not only on the amount and availability of free androgens and estrogens but on their ability to act at the target tissue level. Thus, defects in the androgen receptor or displacement of androgens from their receptors by drugs with antiandrogenic effects (e.g., spironolactone) result in decreased androgen action and, hence, decreased estrogen antagonism at the breast glandular cell level. Finally, the inherent sensitivity of an individual’s breast tissue to estrogen or androgen action may predispose some persons to development of gynecomastia even in the presence of apparently normal concentrations of estrogens and androgens.
ASSOCIATED CONDITIONS
Tables 8-1 and 8-2 list the various conditions and drugs that have been associated with gynecomastia. Although the list is relatively long, almost two-thirds of the patients have either pubertal gynecomastia (approximately 25%), drug-induced gynecomastia (10% to 20%), or no underlying abnormality detected (idiopathic gynecomastia, approximately 25%). Most of the remainder have cirrhosis or malnutrition (8%), primary hypogonadism (8%), testicular tumors (3%), secondary hypogonadism (2%), hyperthyroidism (1.5%), or renal disease (1%) (2). For most pathologic conditions, alterations in the balance between estrogen and androgen levels or action occur through several of the pathophysiologic mechanisms outlined in Table 8-1 and Figure 8-2. One of the best examples is the gynecomastia associated with spironolactone. This aldosterone antagonist inhibits the testicular biosynthesis of testosterone, enhances the conversion of testosterone to the less potent androgen androstenedione, increases the aromatization of testosterone to estradiol, displaces testosterone from SHBG (leading to an increase in its metabolic clearance rate), and binds to the androgen receptors in target tissues, thereby acting as an antiandrogen (7). For an in-depth discussion of the pathophysiology of gynecomastia associated with each of the conditions listed in Tables 8-1 and 8-2, the reader is referred to several reviews (2, 3, 5, 7, 8, 9, 10, 11, 12, 13, 14 and 15).
Only gold members can continue reading. Log In or Register to continue
 Pathology of Benign Breast Disorders
Pathology of Benign Breast Disorders
 Ductal Carcinoma In Situ and Microinvasive Carcinoma
Ductal Carcinoma In Situ and Microinvasive Carcinoma
 Adjuvant Systemic Therapy: Endocrine Therapy
Adjuvant Systemic Therapy: Endocrine Therapy
 Preoperative Endocrine Therapy for Operable Breast Cancer
Preoperative Endocrine Therapy for Operable Breast Cancer



