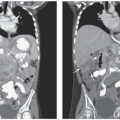
The incidence of lymphoma in children varies by age and varies considerably in different world regions.
1,2 In the United States and in developed countries, malignant lymphoma (including NHL and HL) is the third most common group of malignancies in children after leukemias and brain tumors and account for 15% of all childhood malignancies in children younger than 20 years. NHL is more common than HL in children younger than 10 years, but the relative incidence of HL increases rapidly in children older than 10 years, making the incidence of HL almost twice that of NHL in children between the ages of 15 and 19. Approximately 750 to 800 new cases of childhood NHL are diagnosed in the United States each year. There is a marked male predominance in all age groups, but particularly in children younger than 15 years, in whom three-fourths of the cases occur in males. The incidence of NHL varies considerably by age; NHL is uncommon in children younger than 5 years, accounting for only 3% of cancers; but the incidence of NHL increases steadily throughout life, accounting for 8% to 9% of cancers in children older than 10 years.
2The etiology of NHL is largely unknown. Epidemiologic studies evaluating prenatal and postnatal exposures have not been fruitful for the most part, and exposures studied to date have not been associated with increased risk of lymphoma. The use of pesticides in the home has been linked to the risk of NHL, although no specific agent has been identified.
3 Exposure to drugs and radiation have not been demonstrated to be major risk factors for NHL (except for immunosuppressive drugs—see later and
Chapter 24). Therapy-related secondary NHL in children is rare and is primarily lymphoblastic lymphoma (LBL) or diffuse large B-cell lymphoma (DLBCL).
4 Phenytoin has been associated with “pseudolymphoma,” which usually regresses when the drug is discontinued. Immunodeficiency, whether inherited or acquired, is clearly related to the development of NHL, increasing the incidence of NHL more than 100-fold compared to age-matched controls.
5 The association between Epstein-Barr virus (EBV) and specific NHLs is discussed later.
Initial Evaluation
Unlike adult NHL, which commonly presents as indolent low or intermediate grade, most NHLs in children present as aggressive disseminated disease. Children with NHL benefit from prompt referral to a specialized cancer center for evaluation, staging, and therapy. Potential clinical emergencies in patients with NHL prior to diagnosis include complications from rapidly growing masses: superior/inferior vena cava obstruction, acute airway obstruction, spinal cord compression, pericardial tamponade, intussusception/intestinal obstruction, and central nervous system (CNS) complications. NHL cells frequently have a very high mitotic index, with potential for tumors to grow rapidly and die rapidly, which can result in hyperuricemia and tumor lysis syndrome (TLS). Medical history exploring the infectious history can identify patients at risk for NHL associated with inherited or acquired immune deficiency. Initial studies typically include laboratory studies from peripheral blood to evaluate blood counts, renal function and electrolytes, and lactate dehydrogenase (LDH). If clinically feasible, excisional biopsy is optimal to ensure quality and quantity of tissue for analysis by histology, flow cytometry, cytogenetics, and molecular studies. Once diagnosis is established, radiographic staging of chest, abdomen, and pelvis with a computed tomography (CT) scan is the historical standard, although baseline positron emission tomography (PET)/CT scan may add sensitivity.
6 Bilateral bone marrow aspiration and biopsy and cerebrospinal cytology are also needed for staging.
Staging
The goal of staging studies should be to assess rapidly the extent of disease to determine prognosis and to assign appropriate therapy. Development of a staging system with prognostic utility for children with NHL has been challenging because of the unique biology and differing patterns of spread and response to therapy of the four major subtypes of NHL seen in children and adolescents.
7 The Ann Arbor staging classification
8 does not adequately reflect prognosis in childhood NHL. The progression of disease in childhood NHL does not follow an orderly and predictable pattern of lymphatic spread, as in HL. Extensive extranodal disease is more common in children with NHL than in adults with NHL. Therefore, the clinical staging system proposed at the St. Jude Children’s Research Hospital (
Table 23.1) has been widely accepted.
7 It relies on noninvasive procedures that can be carried out expeditiously. Similar to the Ann Arbor staging system, the St. Jude staging system takes into account the primary site as well as disease extent is considered in assigning clinical stage. The primary differences between St. Jude staging system and the Ann Arbor staging system are as follows. In the St. Jude stage I disease, localized thoracic and abdominal disease are excluded; other localized extranodal disease, however, is considered stage I. In St. Jude stage II disease, localized (resected) abdominal disease is included, and again any thoracic disease is excluded. St. Jude stage III disease includes any thoracic disease, paraspinal disease, or facial nerve palsy in addition to disease on both sides of the diaphragm. Disease involvement in St. Jude system considered stage IV includes marrow or CNS disease, other than paraspinal or facial nerve palsy. On the basis of the St. Jude staging system, almost 40% of children with NHL present with stage I and II and the remainder with more advanced-stage III and IV disease. The distinction between lymphoma and leukemia is arbitrary and is based simply on the percentage of a bone marrow aspirate that is infiltrated by malignant cells. If the bone marrow has more than 25% blasts or malignant cells, the patient is considered to have acute leukemia rather than NHL. Children with between 5% and 25% marrow involvement are considered to have stage IV NHL, whereas those with fewer than 5% blasts in the marrow are considered to not have tumor involved. As opposed to many leukemia criteria, any identifiable tumor cell in the cerebrospinal fluid (CSF) constitutes CNS disease.
The French Society of Pediatric Oncology (SFOP) and Berlin-Frankfurt-Munster (BFM) groups have adapted the St. Jude staging system for treatment assignment for children with B-cell lymphomas (
Table 23.2). The French risk stratification identifies the very best risk patients (Group A) with completely resected localized disease and worst risk patients (Group C) with CNS disease and/or bone marrow involvement. The rest of the patients are included in a very heterogeneous Group B, consisting of patients with all clinical stages I-IV. The BFM risk stratification subdivides patients into four groups based on clinical stage, as well as LDH at diagnosis, a surrogate marker for total disease burden. R1 and R4 are similar to the French Group A and C, while the majority of patients in the French Group B are divided into R2 and R3, depending on LDH level, in the BFM stratifications. For LBL patients, studies have shown that all patients, regardless of stage, have better outcome when treated in a manner similar to that of acute lymphoblastic leukemia (ALL).
9,10 For anaplastic large cell lymphoma (ALCL), the European Intergroup for Childhood NHL (EICNHL) has stratified patients into low-risk versus high-risk based on clinical factors.
11 Patients with one of the following risk factors are considered high-risk: visceral (lung, liver, or spleen), skin, or mediastinal involvement.
12 However, a Children’s Oncology Group (COG) study could confirm only bone marrow involvement as poor prognostic clinical factor.
13With more sensitive techniques, such as flow cytometry or polymerase chain reaction (PCR), several groups have now demonstrated that systemic disease in marrow and blood is much more
frequent than would be predicted by clinical staging systems. Early reports suggest that minimal disseminated disease or minimal residual disease detection in all the major subtypes of pediatric NHL may have prognostic value.
14,15,16,17,18Therapeutic strategies for NHL are based on stage- and histology-directed multiagent chemotherapy, and 5-year survival for children and adolescents is now over 80%. Prognostic factors include age (older children with generally worse outcomes), sites of disease (high stage with worse outcomes than low stage; mediastinal involvement also with inferior outcomes), tumor burden (elevated LDH with inferior outcomes), and response to initial therapy.
NHL Therapy Overview
Prior to 1975, the prognosis for children with NHL was dismal. Most of the children who survived had surgical removal of early-stage disease with or without radiation therapy (RT). The addition of single cytotoxic agents only slightly improved the results with the exception of African children with Burkitt lymphoma (BL). In the original studies of African BL, single-agent chemotherapy was given because radiation was not readily available. Surprisingly, treatment with one or more doses of cyclophosphamide resulted in some cures.
19 Even today about 50% of African BL can be cured with a 28-day course of low-dose cyclophosphamide and prednisone and four intrathecal injections costing less than US$50.
20The morphologic similarity between NHL and ALL and the invariable progression of LBL to a leukemic phase led investigators at St. Jude in the early 1970s to treat NHL with chemotherapy regimens known to be effective in ALL. This novel approach proved to be successful, especially for children with early-stage NHL. However, children with NHL involving the mediastinum had only transient remissions. More intensive ALL regimens such as the Memorial Sloan-Kettering LSA2L2 protocol and the adriamycin (doxorubicin), prednisone, and vincristine (APO) regimen developed at the Dana-Farber Cancer Institute proved to be more successful in children with advanced-stage NHL. During this same time period, Ziegler reported long-term disease control in both American and African patients with BL using a four-drug regimen of cyclophosphamide, methotrexate, vincristine, and prednisone, and Djerassi and Kim demonstrated remissions in children with NHL after moderate-dose methotrexate infusions with citrovorum rescue. The results paved the way for prospective randomized clinical trials comparing leukemia versus lymphoma therapy for NHL in children. These studies provided principles that still hold true today. Leukemia regimens proved to be superior to the lymphoma regimens for LBL and the converse was noted for BL. However, results were roughly equivalent in children with advanced-stage large cell lymphoma.
During the next decade progress continued because of refinements in chemotherapy and advances in supportive care. The 5-year event-free survival (EFS) rates improved to 85% to 95% for children with early-stage NHL and to 70% to 90% for advanced-stage disease. CNS-directed therapy was recognized as an important component of these successful regimens, especially for children with lymphoblastic and Burkitt lymphomas. However, the addition of RT to chemotherapy for both early- and advanced-stage NHL was not shown to improve survival.
9,21,22 Since long-term follow-up studies have shown that the most significant factor for late death following treatment of pediatric NHL is RT, it is currently recommended only for select patients, i.e., treating CNS disease in LBL patients and life-threatening emergencies at diagnosis such as airway compression from a mediastinal mass.
23 The role of surgery in the treatment of children with NHL is mostly limited to diagnostic biopsies, placement of central venous access devices, and treatment of complications of therapy. However, in children presenting with localized disease, e.g., ileocecal intussusception secondary to BL, complete resection of the involved segment of intestine and associated mesentery is recommended. Otherwise, there is no role for performing major tumor resection or debulking procedures in children with NHL.
Another major advancement is the decrease in early death due to TLS. Particular attention to kidney function and to serum levels of uric acid, potassium, calcium, and phosphorus is critical in these children. These patients, particularly those with BL and LBL, are at high risk for TLS and uric acid nephropathy. Measures should be instituted emergently to reduce the likelihood of uric acid nephropathy, including vigorous intravenous hydration (at least twice maintenance), alkalinization of urine with sodium bicarbonate when allopurinol is administered, and careful monitoring of serum electrolytes. Rasburicase, a recombinant urate oxidase, has been shown in recent clinical trials to rapidly lower serum uric acid levels and prevent the metabolic problems associated with tumor lysis, including hyperphosphatemia and renal failure.
24,25 It is not necessary to alkalinize the urine when using rasburicase. Rasburicase should be used in place of allopurinol for children with NHL who have hyperuricemia or who are at high risk for TLS. For the most part, this includes patients with stage III and IV BL and mature B-cell leukemia (primarily Burkitt leukemia), and select patients with bulky LBL and DLBCL. The use of rasburicase has markedly reduced the requirement for dialysis in this population. Additionally, a cytoreduction prophase has been added to many regimens, which also helps achieve tumor control without increasing the risk of clinical deterioration during initiation of therapy, although this does not obviate the need for hydration and uric acid nephropathy prophylaxis.
21,26,27,28
NHL Cellular Classifications: Specific Biological and Clinical Considerations
The groups of lymphomas included under the umbrella of “NHL” are linked by the common feature of not being HL, thus representing a wide range of biological features and clinical presentations. This chapter will focus on the most common forms of NHL in children and adolescents—lymphoblastic lymphoma (precursor T-cell or, less frequently, precursor B-cell), mature B-cell lymphoma (Burkitt, Burkitt-like, DLBCL), anaplastic large cell lymphoma (T-cell or null-cell lymphomas)—and will briefly review mature T-cell lymphomas
29,30 (
Table 23.3).