Abstract
This chapter describes the epidemiology, clinical manifestations, work-up, and management of patients with malignant bone tumors.
Keywords
Osteosarcoma, Ewing sarcoma family of tumors, chondrosarcoma, giant cell tumor of bone, bone tumors, bone cancer, clinical manifestations, epidemiology, treatment, chemotherapy, radiotherapy, surgery
Malignant bone tumors constitute approximately 6% of all childhood malignancies. In the United States, the annual incidence in children under 20 years of age is 8.7 per million. Osteosarcoma (56%), the Ewing sarcoma family of tumors (34%), and chondrosarcoma (6%) comprise the most frequently encountered malignant bone tumors in children and adolescents. In the United States 650–700 children and adolescents under the age of 20 are diagnosed with malignant bone tumors each year.
Figure 27.1 shows the age-specific incidence rate of bone cancers for all races and both sexes.
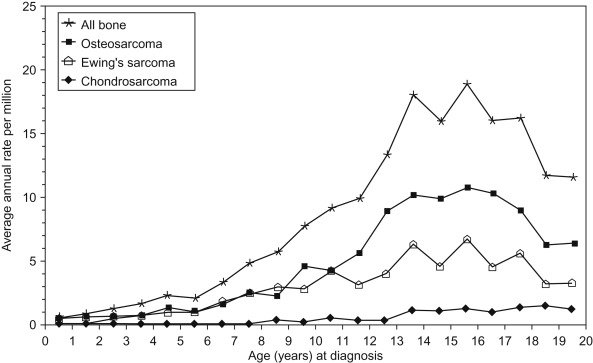
The peak incidence of bone cancer steadily increases through childhood and peaks at age 15. This coincides with the adolescent growth spurt, after which rates decline. Table 27.1 summarizes the differences between osteosarcoma and the Ewing sarcoma family of tumors.
| Osteosarcoma | Ewing sarcoma family of tumors | |
|---|---|---|
| Epidemiology | Bimodal age distribution with peaks in early adolescence and adults over 60 | In children and adolescents, rare in adults |
| Males affected more often than females | Caucasians affected more than other races | |
| African-Americans and other races more affected than Caucasians | ||
| Location | Metaphysis of long bones | Soft tissue mass |
| Flat bones | ||
| Diaphysis of bones | ||
| Metastatic disease | Lung, bone | Lungs, bone, bone marrow |
| Radiographic findings | Radial or “sunburst” pattern | “Permeative” or “moth-eaten” |
| Codman triangle | Bone deposited in “onion peel” appearance | |
| Codman triangle |
Osteosarcoma
Epidemiology
The incidence of osteosarcoma is approximately 800 cases per year in the United States, with approximately 400 cases per year in children less than 20 years old. The incidence is slightly higher in males, and in African-Americans, Asians/Pacific Islanders, and Hispanics than in Whites. Although the etiology of osteosarcoma is unknown, certain factors correlate with occurrence:
- •
Bone growth : The development of osteosarcoma is correlated with increased growth velocity as suggested by:
- •
The peak incidence occurs during the pubescent growth spurt. The age peak is 12 years in girls and 16 years in boys, correlating with the different average ages for pubertal development.
- •
Patients with osteosarcoma are taller than average.
- •
The most common sites are metaphyses of the most rapidly growing bones (distal femur, proximal humerus, proximal tibia).
- •
- •
Genetic factors : Osteosarcoma is a genetically intricate tumor with complex karyotypes, numerous genetic abnormalities, and absence of characteristic chromosomal translocations. Osteosarcoma can be hereditary in some rare cases. Human predisposition syndromes, murine models, genetic analyses of osteosarcoma tumor specimens, and environmental factors may all help to understand the pathogenesis of this disease.
- •
Human predisposition syndromes:
- –
Retinoblastoma —In patients with early-onset bilateral retinoblastoma, who are likely to have germline alteration in the Rb gene, the development of a secondary osteosarcoma or soft-tissue osteosarcoma is independent of the therapeutic modalities or radiation field used in the treatment of the retinoblastoma. Approximately 40% of patients will develop this secondary malignancy by 40 years of age.
- –
P53 —Osteosarcoma is seen frequently in families with Li–Fraumeni syndrome, in which members of the affected families have breast cancer, brain tumors, soft tissue sarcomas, leukemia, adrenocortical carcinoma, and osteosarcoma.
- –
Rothmund–Thomson syndrome —An inherited autosomal recessive disorder consisting of skeletal abnormalities, short stature, underdeveloped or missing forearm bones or thumbs, cataracts, abnormalities of teeth and nails, sparse hair and red, and swollen patches on the skin.
- –
Werner syndrome (also known as progeria adultorum)—A genetic disorder of premature aging consisting of scleroderma-like, thin, wrinkled skin, loss of subcutaneous fat, graying and loss of hair, bilateral cataracts, hypogonadism, and premature menopause.
- –
Diamond–Blackfan anemia —A rare congenital bone marrow failure disorder characterized by pure red cell aplasia. About 50% of patients have mutations in the ribosomal protein subunit genes that are associated with increased incidences of osteosarcoma among other malignancies.
- –
- •
Murine models that produce osteosarcoma include those with alterations in the p53, Rb, Wnt, Notch, IGF, mTor pathways, MYC , FOS as well as chronic parathyroid hormone exposure.
- •
Genetic analyses of osteosarcoma tumor specimens:
- –
Osteosarcomas are genetically complex with numerous alterations in each tumor sample. These include a multitude of genes involved in cell cycle regulation, differentiation, oncogenic transformation, cell signaling, signal transduction, among many pathways. Consistent chromosomal gains and losses occur at genetic sites in which the gene(s) of interest have not been clearly identified.
- –
Approximately 3% of patients with spontaneous osteosarcoma are found to have a germline alteration in p53 (Li–Fraumeni syndrome).
- –
There are non-germline mutations of both p53 and Rb in osteosarcoma tumors. Twenty-five percent have point mutations in the p53 gene, 10–20% have p53 rearrangements, 75% have loss of one 17q allele, and 60% of OS tumors have a loss of heterozygosity at 13q of the site of the Rb gene.
- –
- •
- •
Environmental factors : The environmental factor most consistently associated with the development of osteosarcoma is radiation therapy. Bone turnover also may be related to osteosarcoma development. The evidence for these is:
- •
Osteosarcoma may arise in any previously irradiated bone.
- •
The interval between irradiation and the appearance of osteosarcoma has ranged from 4 to 40 years but is generally 10–20 years, suggesting it is not a major etiologic factor in the younger patients.
- •
Paget disease of bone and other disorders associated with rapid bone turnover are associated with a higher incidence of osteosarcoma.
- •
Pathology
The histologic diagnosis of osteosarcoma is dependent on the demonstration of malignant osteoid. Table 27.2 lists the classification of osteosarcoma. Although several of the rarer variants may be confused radiographically or histologically with other entities, the pathologic hallmark of osteosarcoma is the production of osteoid. Parosteal osteosarcoma is generally considered a low-grade variant of osteosarcoma and is treated with surgical resection alone.
|
Clinical Manifestations
Symptoms are usually present for several months before the diagnosis is made, with an average duration of approximately 3 months. Patients commonly present with the following symptoms:
- •
Pain at site of tumor (90%)
- •
Local swelling (50%)
- •
Decreased range of motion (45%)
- •
Pathologic fracture (8%)
- •
Joint effusion can occur rarely. Its presence may suggest an extension of the tumor into the joint space.
It is important to be aware of the sites and ages at which osteosarcoma presents as this helps distinguish it from other entities which occur more commonly in this population, such as growing pains and osteomyelitis. Osteosarcoma can arise in any bone but has a propensity for the metaphyseal portions of long bones, particularly in the lower extremity around the knee ( > 50% involving distal femur or proximal tibia). The most common presenting symptom is persistent pain and swelling at the primary site. Approximately 10–20% have evidence of metastatic disease at the time of diagnosis, with lungs and bone being the most common sites. Systemic manifestations of osteosarcoma are rare at initial presentation, and when they occur they are usually associated with significant metastatic disease.
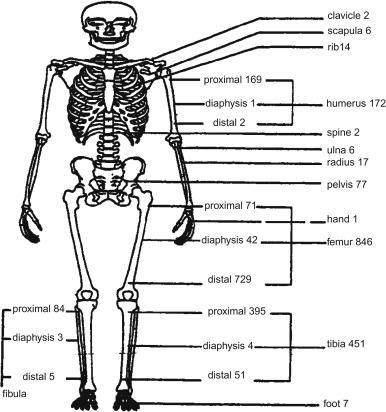
Figure 27.2 shows the skeletal distribution of patients with osteosarcoma. The most common primary sites in individuals less than 25 years of age are:
- •
Lower long bones (74.5%)
- •
Upper long bones (11.2%)
- •
Pelvic region (3.6%)
- •
Face or skull (3.2%)
- •
Mandible (1.9%)
- •
Chest region (1.8%)
- •
Vertebral column (1.2%)
- •
Lower short bones (1.1%)
- •
Upper short bones (0.3%).
Diagnostic Evaluation
The diagnostic evaluation should delineate the extent of the primary tumor and the presence or absence of metastatic disease. All patients should have the following evaluation:
- •
History .
- •
Physical examination .
- •
Blood count .
- •
Urinalysis .
- •
Blood urea nitrogen (BUN), creatinine, liver enzymes, alkaline phosphatase, total bilirubin, and lactate dehydrogenase : 30–50% of patients have elevated alkaline phosphatase which has been associated with a worse prognosis.
- •
Radiographs of the entire affected bone —The Codman triangle, the formation of new periosteum and the elevation of the cortex, may be present.
- •
Computed tomography (CT) and magnetic resonance imaging (MRI) of the entire affected bone . Imaging of the primary tumor includes MRI, because the extent of soft tissue involvement, especially that of neurovascular structures, will dictate the feasibility of limb salvage surgery. MRI will also show the extent of marrow involvement, essential information in surgical planning.
- •
Technetium-99 bone scan and/or whole body 18-fluoro-deoxyglucose positron emission tomography (FDG PET)/CT scan— Approximately 10% of patients will have evidence of bone metastases on bone scan. Whole-body FDG PET/CT is used for diagnostic evaluation and may be more sensitive and specific for diagnosing bone metastases when compared to technetium-99 bone scan. Bone scan, however, is still considered standard for detection of osseous metastases.
- •
High-resolution chest CT —The use of chest CT is essential for staging patients, because it detects metastases not seen on chest radiographs. If a lesion detected on chest CT cannot be diagnosed as metastatic disease, histologic confirmation is indicated.
- •
Assessment of renal function —Institutional practice varies with some obtaining a creatinine clearance or radionuclide GFR study to assess renal function prior to chemotherapy whereas others rely solely on the serum creatinine with a calculated estimate of glomerular filtration rate.
- •
Echocardiogram to assess cardiac function prior to chemotherapy.
- •
Baseline audiogram prior to chemotherapy.
- •
Fertility preservation —sperm banking should be offered whenever feasible. If timing allows, oocyte cryopreservation is the standard of care for postpubertal young women who are not prepared to undertake embryo cryopreservation. Cryopreservation of ovaries has not become a clinical standard at present but interest in this approach as a means of preserving fertility continues to increase.
- •
Biopsy of primary site and possibly secondary sites if questionable for other diagnosis —Biopsy and pathologic review are essential to establish diagnosis of osteosarcoma. Biopsy should be performed by an orthopedic surgeon, radiologist, or interventional radiologist with expertise. Improperly performed biopsies can contaminate unaffected tissue and alter definitive surgical resection. Either an open biopsy or image-guided percutaneous core biopsy can be performed. The biopsy should be performed with future surgery in mind as a poorly placed biopsy may jeopardize future limb salvage surgery.
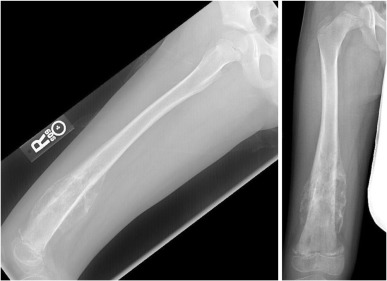
Radiographically, osteosarcoma typically shows destruction of normal trabecular bone in the metaphysis of the affected long bone along with the following findings ( Figure 27.3 ):
- •
Soft-tissue extension (75%)
- •
Radiating calcification, or sunburst (60%)
- •
Osteosclerotic lesion (45%)
- •
Lytic lesion (30%)
- •
Mixed lesion (25%).
Treatment of Localized Osteosarcoma
Treatment relies on early and proper diagnosis, neoadjuvant and adjuvant multiagent chemotherapy, and aggressive surgery, with limb-preserving therapy when possible. The tumor is generally viewed as radioresistant and radiation therapy only plays a role in initial management in a case-by-case basis when positive margins are unable to resect. The survival rate of non-metastatic osteosarcoma is approximately 70%.
Surgery
The surgical approach to osteosarcoma includes surgical biopsy, followed by either limb salvage surgery or amputation. Complete resection is a prerequisite for cure.
Surgery should remove all gross and microscopic tumor with a margin of normal tissue to prevent local recurrence as positive margins and poor necrosis lead to a higher risk of recurrence. Tumor involvement of neurovascular structures, seeding of surrounding compartments from a pathological fracture, and inadequacy of surrounding soft tissue for closure are the common contraindications for limb salvage. In recent decades, approximately 85% of patients with osteosarcoma have been able to be surgically managed with limb salvage surgery. If adequate surgical margins are achieved, the rate of local recurrence and survival after amputation and limb salvage surgery are equivalent.
Definitive surgery is generally performed following preoperative (neoadjuvant) chemotherapy. Delaying definitive surgery until after neoadjuvant chemotherapy is the standard treatment approach, though there has been no defined survival benefit.
Neoadjuvant chemotherapy offers several theoretical advantages:
- •
Decrease in tumor-related edema and shrinkage of the tumor at the primary site so as to facilitate less radical surgery.
- •
Initial chemotherapy directed toward the assumed micrometastases which are present in 80–90% of patients.
- •
Assessment of sensitivity of primary tumor to chemotherapy which has prognostic significance. Greater than 90% necrosis is considered a good response and portends an improvement in overall survival.
The type of surgery is determined by tumor location, size, extramedullary extent, the presence of metastatic disease, age, skeletal development, and the patient’s lifestyle choices. If complete excision cannot be accomplished with limb-sparing surgery, amputation is indicated. Guidelines for the use of limb-sparing surgery include:
- •
No major neurovascular involvement by tumor.
- •
Ability to have a wide resection of affected bone with a normal muscle cuff in all directions.
- •
Ability to perform en bloc removal of all previous biopsy sites and all potentially contaminated tissue.
- •
Adequate motor reconstruction.
Chemotherapy
The use of chemotherapy results in significant improvement in disease-free survival. More than 80–90% of patients treated with surgery alone will develop metastatic disease. The standard components of multiagent chemotherapy for osteosarcoma are high-dose methotrexate (HDMTX), doxorubicin (Adriamycin, ADM), and cisplatin (CDDP), a regimen often referred to as MAP. Figure 27.4A shows the schedule and doses of MAP regimens.
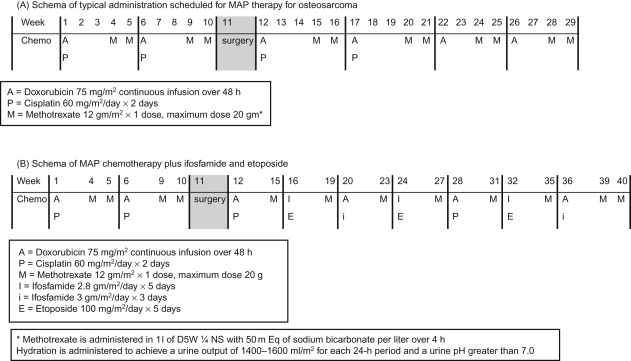
Ifosfamide (IFO) and etoposide (VP16) are other agents with demonstrated activity in osteosarcoma, however, the use of these agents upfront in the management of localized osteosarcoma has not shown a survival advantage at this time.
Specific HDMTX administration guidelines must be followed to prevent renal injury and life-threatening toxicity. Normal renal function should be documented prior to administration. Alkalinizing intravenous fluids should be administered with HDMTX and continued until a sufficient decrease in MTX blood levels has occurred. MTX levels should be monitored starting 4 h after the beginning of MTX administration and then every 24 h from the starting time of the MTX infusion. Leucovorin should be initiated 24 h after HDMTX administration in a dose of 15 mg/m 2 oral or IV route every 6 h until the MTX level is less than 1.0×10 7 mol/l. Patients whose MTX excretion is delayed require an increased dose or duration of leucovorin, according to Table 27.3 . Glucarpidase (Voraxaze) is now FDA-approved for patients with toxic plasma methotrexate levels in patients with delayed clearance due to renal dysfunction. It should be administered at 50 units/kg IV once.
| Expected excretion | Delayed excretion | Grade I toxicity (mild) | Grade II toxicity (moderate) | Grade III toxicity (severe) | Grade IV toxicity (life-threatening) | |
|---|---|---|---|---|---|---|
| 24 | ≤10 µM (1×10 −5 M) | >10 to <50 μM (1–5×10 −5 M) | >10 to <50 μM (1–5×10 −5 M) | ≥50 μM to <500 μM (5–50×10 −5 M) | ≥500 μM (5×10 −4 M) | |
| 48 | ≤1 µM (1×10 −6 M) | ≥1 μM to <5 μM (1–5×10 −6 M) | ≥1 μM to <5 μM (1–5×10 −6 M) | ≥5 μM to <100 μM (5×10 −6 M) –(1×10 −4 M) | ≥100 μM (1×10 −4 M) | |
| 72 | ≤0.1 µM (1×10 −7 M) | ≥0.1 to <0.5 μM (1–5×10 −7 M) | ≥0.5 to <5 μM (0.5–5×10 −6 M) | ≥0.5 to <5 μM (0.5–5×10 −6 M) | ≥5 to <50 μM (0.5–5×10 −5 M) | ≥50 μM (5×10 −5 M) |
| Increase in baseline creatinine a | 25–50% | 50–100% | >100% | |||
| Other a | Grade I–II stomatitis | On previous MTX course: Grade III–IV stomatitis, myelosuppression | ||||
| Leucovorin | 15 mg/m 2 q6h PO/IV Until MTX level <0.1 μM (1×10 −7 M) | 15 mg/m 2 q6h PO/IV | 15 mg/m 2 q6h PO/IV b | 15 mg/m 2 q3h IV Until MTX level b | 150 mg/m 2 q3h IV Until MTX level b | 1500 mg/m 2 q6h IV (maximum dose 1500 mg) b |
| Recheck at 96 h if <0.08 μM (8×10 −8 M) discontinue leucovorin if ≥0.08 μM discontinue leucovorin when level <0.05 μM (5×10–8 M) | ||||||
| Other interventions | Hydration 125 ml/m 2 /h | Hydration 125 ml/m 2 /h | Increase hydration 200 ml/m 2 /h | Increase hydration 200 ml/m 2 /h | Consider glucarpidase increase hydration 200 ml/m 2 /h | Consider glucarpidase Increase hydration 200 ml/m 2 /h |
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


