Acute Lymphoblastic Leukemia
Leukaemia Research Cytogenetics Group, Northern Institute for Cancer Research, Newcastle University, Level 5 Sir James Spence Institute, Royal Victoria Infirmary, Newcastle-upon-Tyne NE1 4LP, UK
Keywords
• Acute lymphoblastic leukemia • Childhood • Adult • Diagnosis • Prognosis • Genetics
ALL is classified broadly as B-lineage and T-lineage.1 B cell precursor ALL (BCP-ALL) is a malignancy of the lymphoblasts committed to the B cell lineage. It is generally associated with a good outcome in children with a cure rate of approximately 85%, whereas in adults the overall survival is less than 50%. Those features associated with an adverse outcome in BCP-ALL are cytogenetics, infancy, age under 10 years, high white blood cell count, slow response to initial therapy, and the presence of minimal residual disease after first therapy.
T-lineage ALL (T-ALL) is a malignancy of the thymocytes that accounts for approximately 15% of childhood and 25% of adult ALL. It is most common in adolescents and more frequent in males than females. T-ALL is generally considered to be an aggressive high-risk disease. In these patients the presence of minimal residual disease is a strong prognostic factor. Recently, intensified treatment has improved outcome of T-ALL patients.2
Many important chromosomal abnormalities have been reported in both BCP-ALL and T-ALL. It is of interest that a number have been shown to arise prenatally, long before the onset of leukemia is diagnosed.3 In childhood ALL, the incidences of individual chromosomal abnormalities are well-established (Fig. 1). It is also known that their distribution varies according to age (Fig. 2).4 Especially in BCP-ALL, these chromosomal abnormalities are strong independent indicators of risk of relapse,5 whereas in T-ALL they contribute significantly to the understanding of the biology of the disease. These genetic features provide the focus for this article in which those changes with the greatest significance in relation to their biological and clinical relevance are presented. The main abnormalities and their associated genes are summarized for BCP-ALL in Table 1 and T-ALL in Table 2. For a detailed catalog of chromosomal abnormalities in ALL, the reader is referred to Harrison and Johansson.6 In the author’s opinion, within the current era, the definition of cytogenetics, at least for ALL, must be inclusive of genomic abnormalities detected at the DNA level.
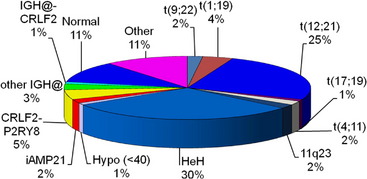
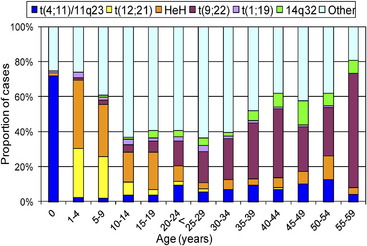
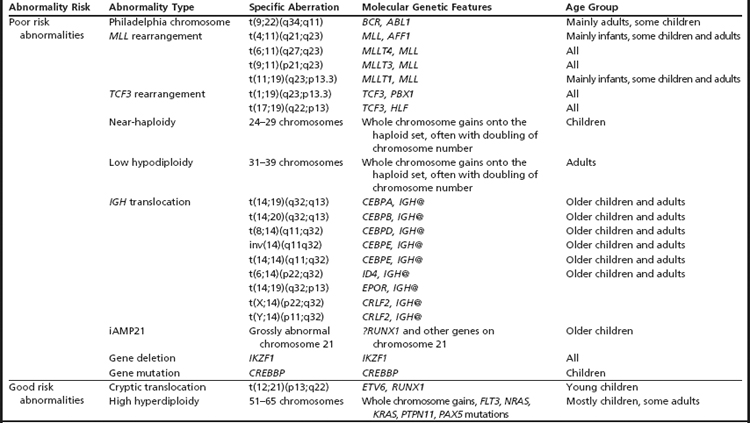
Table 2 Significant genetic abnormalities in T-ALL
| Type of Aberration | Aberration | Molecular Genetic Features |
| Aberrant expression of transcription factors and related genes | t(1;7)(p34;q34) | LCK, TRB@ |
| TAL1 deletion | TAL1, STIL | |
| t(6;7)(q23;q34 | MYB, TRB@ | |
| t(7;9)(q34;q32) | TAL2, TRB@ | |
| t(7;9)(q34;q34.3) | NOTCH1, TRB@ | |
| t(7;11)(q34;p13) | LMO1, TRB@ | |
| t(7;11)(q34;p15) | LMO2, TRB@ | |
| t(7;12)(q34;p13.3) | CCND2, TRB@ | |
| t(7;19)(q34;p13) | LYL1, TRB@ | |
| t(8;14)(q24;q11) | MYC, TCRA/D@ | |
| t(11;14)(p13;q11) | LMO1, TRA@/TRD@ | |
| t(11;14)(p15;q11) | LMO2, TRA@/TRD@ | |
| t(12;14)(p13;q11) | CCND2, TRA@. | |
| inv(14)(q11q32) | BCL11B, TRD@ | |
| t(14;14)(q11;q32) | BCL11B, TRD@ | |
| NKX2-1 rearrangements | NKX2-1 | |
| NKX2-2 rearrangements | NKX2-2 | |
| MEF2C rearrangements | MEF2C | |
| t(14;21)(q11;q22) | OLIG2, TRA@ | |
| Abnormalities of homeodomain genes | t(7;10)(q34;q24) | TLX1, TRB@ |
| t(10;14)(q24;q11) | TLX1, TRA@/TRD@ | |
| t(5;14)(q35;q32) | TLX3, BCL11B | |
| Abnormalities of the HOXA cluster | inv(7)(p15q34) | HOXA@, TRB@ |
| t(7;7)(p15;q34) | HOXA@, TRB@ | |
| t(7;14)(p15;q11) | HOXA@, TRD@ | |
| t(7;14)(p15;q32) | HOXA@, BCL11B | |
| Fusion transcripts | t(6;11)(q27;q23) | MLLT4, MLL |
| t(9;9)(q34;q34) | NUP214, ABL1 | |
| t(9;14)(q34;q32) | EML1, ABL1 | |
| t(10;11)(p12;q14) | PICALM, MLLT10 | |
| Copy number changes | MYB duplication | MYB |
| del(9p) | CDKN2A | |
| del(18)(p11) | PTPN2 | |
| Mutations | NOTCH1 mutations | NOTCH1 |
| FBXW7 mutations | FBXW7 | |
| PFH6 mutations | PFH6 |
B Cell Precursor ALL
Poor Risk Abnormalities
Philadelphia chromosome
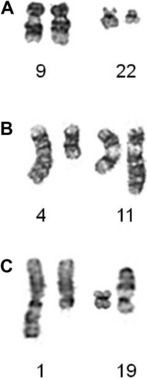
Particularly in childhood BCP-ALL, chromosomal abnormalities are important for risk stratification for treatment. The most well-known is the translocation between chromosomes 9 and 22, t(9;22)(q34;q11), otherwise known as the Philadelphia chromosome (Ph), which is the hallmark chromosomal change of chronic myeloid leukemia (Fig. 3A). The translocation gives rise to the BCR-ABL1 fusion with constitutive activation of the tyrosine kinase, BCR-ABL1. The translocation accounts for approximately 2% of BCP-ALL in children and has been associated with a dismal outcome. More recently, treatment with the tyrosine kinase inhibitor imatinib has significantly increased disease-free survival, although it is as yet too early to know if this result will translate into improvements in overall survival.7 Trials are in progress to determine whether the new-generation tyrosine kinases, for example dasatinib and nilotinib, can further improve outcome.
MLL rearrangements
Chromosomal abnormalities involving the chromosomal band 11q23, resulting in rearrangements of the MLL gene, account for approximately 2% of childhood BCP-ALL. The translocation, t(4;11)(q21;q23) giving rise to the MLL-AFF1 fusion (previously known as MLL-AF4), is the most common (Fig. 3B). However, more than 80 MLL partners have been reported, with more than 50 of them characterized at the molecular level.8 Childhood BCP-ALL patients with t(4;11) have also been classified as high-risk, although this statistic is currently under review because the outcome seems to be age-dependent.5
TCF3 rearrangements
Among patients with TCF3 rearrangements, those with t(1;19)(q23;p13.3)/TCF3-PBX1 fusion (Fig. 3C) were originally regarded as high-risk on some treatment protocols. However, on modern therapy they are classified as standard risk.9 In contrast, the rare variant, t(17;19)(q22;p13)/HLF-TCF3 fusion, has a dismal outcome on all therapies.10 Thus, accurate identification is important for appropriate risk stratification.
Near-haploidy and low hypodiploidy
Near-haploidy (24–29 chromosomes) and low hypodiploidy (31–39 chromosomes) are rare abnormalities comprising less than 1% each of childhood BCP-ALL. These abnormalities are associated with a poor treatment response, and patients who have them are stratified as high-risk. The abnormalities represent numerical chromosomal changes in ploidy level characterized by the gain of specific chromosomes onto the haploid chromosome set. In the majority of patients, a population of cells with an exact doubling of this chromosome number is present, producing tetrasomies of the gained chromosomes.11,12 The doubling of a near-haploid population results in a karyotype with over 50 chromosomes, which is easily mistaken for high hyperdiploidy (see high hyperdiploidy section below). The presence of tetrasomies rather than trisomies of the gained chromosomes distinguishes near-haploid doubling from the classical form of high hyperdiploidy. It is important that these differences are identified because of the poor outcome of the near-haploid doubling compared with the good prognosis associated with high hyperdiploidy.
IGH@ Translocations
Translocations involving the immunoglobulin heavy chain locus, IGH@, at 14q32 with a range of partner genes are emerging as a significant subgroup in childhood BCP-ALL.13–16 The partners include five of the CEBP gene family members13 and the cytokine receptors: EPOR and CRLF2.15,16 The latter is a cryptic translocation involving IGH@ and the CRLF2 gene, which is located within the pseudoautosomal region (PAR1) of both sex chromosomes, t(X;14)(p22;q32) or t(Y;14)(p11;q32).16 All IGH@ translocations occur more frequently in older children and adolescents and, although numbers are small, these translocations seem to have an inferior outcome.
iAMP21
The cytogenetic subgroup, intrachromosomal amplification of chromosome 21 (iAMP21), was identified during routine screening for the presence of the ETV6-RUNX1 fusion by fluorescence in situ hybridization (FISH).17,18 Patients are negative for the ETV6-RUNX1 fusion but show multiple copies of RUNX1 (3 or more additional signals). In metaphase, multiple signals are seen in tandem duplication along an abnormal chromosome 21.19 In interphase, the signals are clustered together. Cytogenetics, FISH, and genomic approaches have shown that the morphology of the abnormal chromosome 21 is highly variable between patients (Fig. 4). The commonly amplified region always includes the RUNX1 gene.19–21 More recent studies have confirmed that iAMP21 is a primary genetic change and that the mechanisms leading to instability of the abnormal chromosome 21 are inherent within the chromosome structure.22
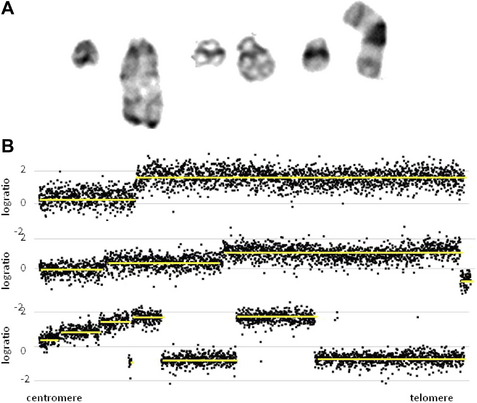
iAMP21 was originally described as poor risk,23 although the outcome has since been shown to be protocol-dependent.24 Thus its accurate detection is important in order to guide therapy.
Good Risk Abnormalities
ETV6-RUNX1 fusion
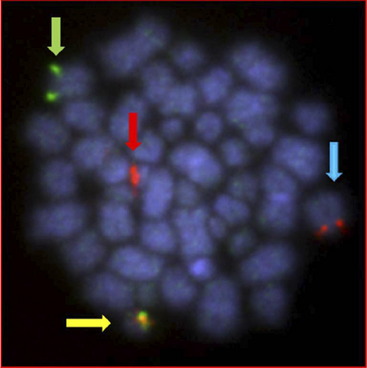
A significant structural abnormality is the cryptic translocation, t(12;21)(p13;q22)/ETV6-RUNX1 fusion (Fig. 5). This abnormality occurs in approximately 25% of younger children with BCP-ALL.25 These patients have an extremely good prognosis.
High hyperdiploidy
High hyperdiploidy (51–65 chromosomes) is an important numerical abnormality.26 It accounts for approximately 30% of childhood BCP-ALL and is characterized by the gain of specific chromosomes. High hyperdiploidy is also associated with an excellent outcome in children. It is largely unknown why both abnormalities are linked to a good prognosis. The reasons may be in part age-related because, overall, younger children have the best treatment response, and these abnormalities occur predominantly in younger children.
Submicroscopic Abnormalities
A significant discovery was that the disruption of genes involved in B cell development, through deletions, amplifications, point mutations, and structural rearrangements, played an important role in leukemogenesis in childhood BCP-ALL.27 Approximately 40% of these patients had abnormalities of genes involved in the B cell development and differentiation pathway, including: PAX5, TCF3, EBF1, LEF1, IKZF1 and IKZF3. Other genes frequently affected were those controlling cell cycle progression including: CDKN2A, CDKN1B, and RB1 (Fig. 6A).28,29 Whether there is a link between these abnormalities and outcome has become a critical question. Alterations of IKZF1 (which encodes the lymphoid transcription factor, IKAROS) are common in BCR-ABL1-positive ALL, known to have a poor outcome.30 However, IKZF1 deletions have also been found to be a marker of poor prognosis in Ph-negative BCP-ALL patients.31–33 Currently this observation is being further validated in prospective independent and unselected trial-based patient cohorts.
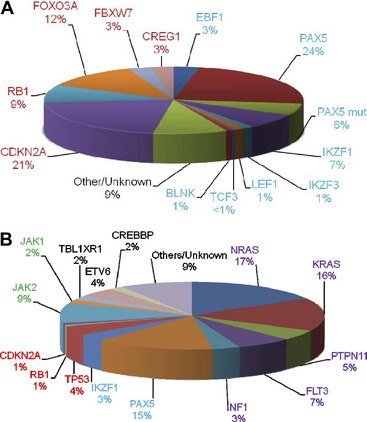
Fig. 6 The relative incidences of (A) deletions and (B) mutations in the key signaling pathways in childhood BCP-ALL. The genes are color-coded according to the pathway to which they belong: B cell differentiation and development, blue; TP53/RB1, red; Ras signaling, purple; JAK/STAT, green, noncanonical pathways and other/unknown genes, black. (A) Deletions are shown for the B cell differentiation and development and TP53/RB1 pathways only. The PAX5 mutation rate stated by Mullighan and colleagues27 is given (PAX5 mut). The relative incidences of the mutations in the lower chart are estimated from Zhang and colleagues.42
Related posts:
 Cytogenetics
Cytogenetics
 Structural Rearrangements: Detection and Elucidation of Mechanisms Using Cytogenomic Technologies
Structural Rearrangements: Detection and Elucidation of Mechanisms Using Cytogenomic Technologies
 Evolving Picture of Microdeletion/Microduplication Syndromes in the Age of Microarray Analysis: Variable Expressivity and Genomic Complexity
Evolving Picture of Microdeletion/Microduplication Syndromes in the Age of Microarray Analysis: Variable Expressivity and Genomic Complexity
 In Situ Hybridization
In Situ Hybridization
 Syndromes
Syndromes
 Myeloid Leukemia: Conventional Cytogenetics, FISH, and Moleculocentric Methodologies
Myeloid Leukemia: Conventional Cytogenetics, FISH, and Moleculocentric Methodologies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


