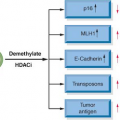
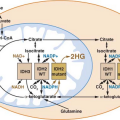
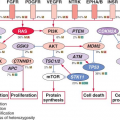
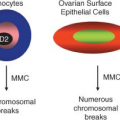
Chromosomal aberrations in tumors are regarded traditionally as evidence of gene deregulation and genome instability, and their detection may facilitate the identification of crucial genes and regulatory pathways that are perturbed in diseases. Several powerful analytical tools are currently available for analyzing chromosomal aberrations (reviewed in
ref. 9). Comparative genomic hybridization (CGH), in particular array CGH, enables high-throughput analysis of DNA copy number and yields comprehensive information applicable to determining the molecular pathogenesis of human HCC. Meta-analysis of CGH studies of chromosome aberrations in human HCC shows that specific chromosomal gains and losses correlate with etiology and histologic grade (
Table 18.1).
10 In HCC the most frequent amplifications of genomic material involve 1q (57.1%), 8q (46.6%), 6p (22.3%), and 17q (22.2%), whereas losses are most common in 8p (38%), 16q (35.9%), 4q (34.3%), 17p (32.1%), and 13q (26.2%). Deletions of 4q, 16q, 13q, and 8p correlate with HBV infection and lack of HCV infection. Chromosomes 13q and 4q are significantly underrepresented in poorly differentiated HCC, and gains of 1q correlate with other high-frequency alterations.
11 Amplifications and deletions often occur on chromosome arms at sites of oncogenes (e.g.,
MYC on 8q24) and tumor suppressor genes (e.g.,
RB1 on 13q14), as well as at several loci that contain genes with known and/or
suspected oncogenic functions (e.g.,
FZD3, WISP1, SIAH-1, and
AXIN2, all of which modulate the WNT signaling pathway). In this meta-analysis, etiology and poor differentiation of HCC correlated with specific genomic alterations. In preneoplastic dysplastic nodules (DNs), amplifications are most frequent in 1q and 8q, whereas deletions occur in 8p, 17p, 5p, 13q, 14q, and 16q.
11 Gain of 1q appears to be an early event developing in DN, possibly predisposing affected cells to acquire additional chromosomal aberrations.
Bioinformatic analysis of CGH data was recently used to develop a progression model for human HCC.
11 Based on an evolutionary tree constructed from statistically significant CGH events, three subgroups of patients with different patterns of HCC progression were identified. The subgroups reflect the extent of tumor progression as indicated by the number of chromosomal aberrations, tumor stage, tumor size, and disease outcome. Gains of 1q21-23 and 8q22-24 appear to be early genomic events in development of HCC and gain of 3q22-24 a late genomic event, the latter associated with tumor recurrence and poor survival. The HCC progression model uncovered chromosomal imbalances associated with clinical pathologic characteristics of the disease and explained a significant part of the variations in clinical outcome among the HCC patients.
11These two studies illustrate the power of CGH analysis to identify the functional significance of genomic alterations in human HCC. Nevertheless, because CGH only analyzes genomic DNA, additional studies are required to measure and integrate data on global gene expression to confirm the roles of candidate genes. This can be accomplished by adapting the expression imbalance
map method and array CGH analysis (reviewed in
ref. 12). This approach was recently applied to human HCC.
13 Using regional pattern recognition approaches, the authors discovered the most probable copy number-dependent regions and 50 potential driver genes (
Table 18.2). At each step of the gene selection process, the functional relevance of the selected genes was evaluated by estimating the prognostic significance of the selected genes. Further validation using small interference RNA-mediated knockdown experiments showed proof-of-principle evidence for the potential driver roles of the genes in HCC progression (i.e.,
NCSTN and
SCRIB). In addition, systemic prediction of drug responses implicated the association of the 50 genes with specific signaling molecules (
mTOR, AMPK, and
EGFR). It was concluded that the application of an unbiased and integrative analysis of multidimensional genomic data sets can effectively screen for potential driver genes and provides novel mechanistic and clinical insights into the pathobiology of HCC.
It seems inevitable that new and improved array designs for both CGH and gene expression and the advent of whole genome sequencing combined with better software for statistical analysis of the data will continue to emerge.