Background
Two of the most abundant lipid components in plasma are cholesterol and triglycerides (TG); however, both are virtually insoluble in water. To be transported to various tissues, these lipids must be packaged into a lipoprotein particle, the core of which is composed of cholesteryl esters and TG, and the surface is composed of phospholipids, free cholesterol, and apolipoproteins . Lipoprotein particles are freely water soluble and along with albumin are the main transporters of hydrophobic substances in plasma.
In man and most other mammals, there are four major lipoprotein classes ( Table 9.1 ) . Chylomicrons (CM) and very low-density lipoproteins (VLDL) are the main TG-carrying particles. Low-density lipoproteins (LDL), which are produced after the lipolysis of TG on VLDL, are cholesterol-rich lipoproteins. High-density lipoproteins (HDL) are also cholesterol-enriched and besides apolipoprotein (apo) A-I (apoA-I), its main structural protein, they transport a wide variety of proteins such as apoA-II or apoC-II. It is important to note that all the different lipoproteins contain the same lipids but only in different proportions ( Table 9.1 ) and they are commonly classified by either their density or electrophoretic mobility [origin (CM), pre-β (VLDL), β (LDL), and α (HDL)].
| CM | VLDL | LDL | HDL | |
|---|---|---|---|---|
| Origin | Gut | Liver | VLDL | Liver, intestine |
| Density (g/mL) | 0.94 | 0.94–1.006 | 1.019–1.063 | 1.063–1.21 |
| Electrophoretic mobility | Origin | Pre-β | β | α |
| Major proteins | apoB-48, C, E | apoB-100, C, E | apoB-100 | apoA-I, A-II, C, E |
| % Protein | 1% | 10% | 20% | 50% |
| Major lipids | TG (90%) | TG (55%) | Cholesterol (50%) | Phospholipids (25%), cholesterol (20%) |
| Main function | Transport dietary TG | Transport endogenous TG | Transport cholesterol esters | Reverse cholesterol transport |
As will be discussed in more detail later, disturbances in lipoprotein metabolism have two major pathophysiologic consequences. Elevated LDL-cholesterol (LDL-C) can predispose to premature atherosclerosis , whereas very high concentrations of plasma TG (>1000 mg/dL) carried by CM and VLDL can precipitate pancreatitis . It is unclear how HDL relates to the pathogenesis of atherosclerosis, but they are at the very least a useful negative biomarker for coronary heart disease (CHD) risk.
Overall, lipoprotein metabolism can be viewed as three interconnected pathways ( Fig. 9.1 ), namely the exogenous, endogenous, and reverse cholesterol transport pathways . The exogenous pathway ( Fig. 9.1 , pathway I) is involved in the absorption of dietary fat and the delivery of TG to peripheral cells and the liver. In this pathway, dietary fat is first packaged in the intestine as a CM particle, which contains apoB-48 and is initially secreted into lymphatics, which eventually drain into the circulatory system. TG in CM are hydrolyzed by a pivotal enzyme in lipoprotein metabolism, lipoprotein lipase (LPL), which is bound to the vessel wall and requires apoC-II as its cofactor. The hydrolysis of TG results in the release of fatty acids, which are avidly taken up by peripheral tissues and can be re-esterified into TG for energy storage or can undergo β-oxidation for energy production. The CM remnants that are formed as a consequence of lipolysis by LPL are rapidly removed by the liver by LDL receptor-related protein (LRP) and by proteoglycans in the hepatic sinusoidal spaces.
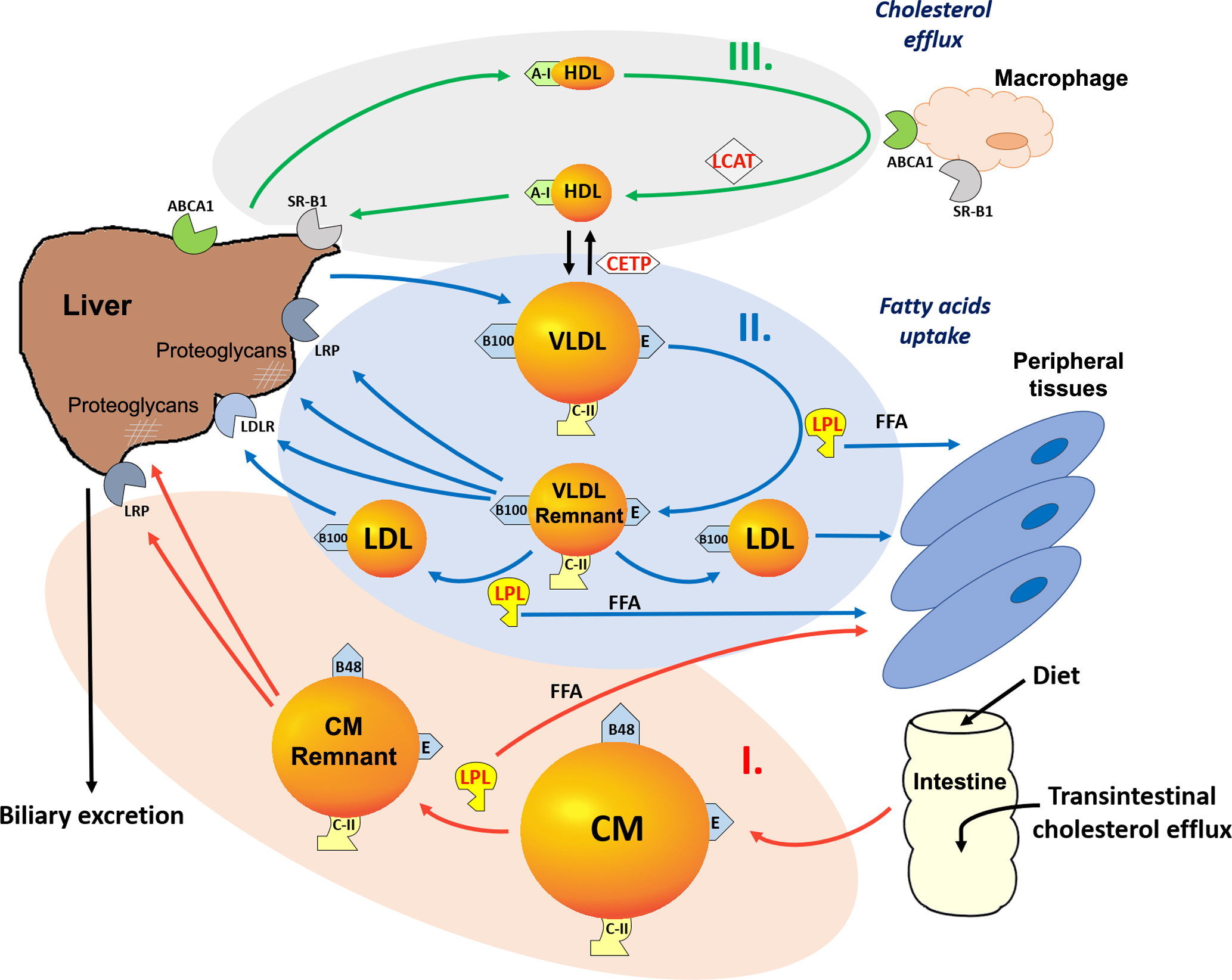
The endogenous pathway ( Fig. 9.1 , pathway II) is responsible for the delivery of hepatic-derived TG as a source of energy to peripheral cells, during fasting. In this pathway, VLDL, which contain apoB-100 and are enriched in TG, are secreted by the liver. Upon entry into the circulation, VLDL have the same fate as CM in that they are acted upon by LPL, resulting in the formation of VLDL remnants, which are also called intermediate-density lipoproteins (IDL). VLDL also become enriched in cholesteryl esters, because of cholesteryl ester transfer protein (CETP)-mediated exchange of lipids between VLDL and HDL. VLDL remnants can be directly removed by the liver by LDL receptors (LDLR), LRP and proteoglycans or can continue to lose additional TG by LPL-dependent lipolysis to generate LDL. LDL can be removed from the circulation by the LDLR, which are on many cell types, but it is the level of LDLR on the liver that primarily regulates plasma levels of LDL. LDL are atherogenic most likely because of their propensity to be oxidized and to be taken up by macrophages in the vessel wall, which results in the formation of foam cells and fatty streaks, one of the earliest hallmarks of atherosclerosis .
In contrast to the apoB-containing lipoproteins, which primarily promote the delivery of TG to peripheral tissues, HDL participate in the reverse cholesterol transport pathway ( Fig. 9.1 , pathway III). This is the pathway by which cells maintain cholesterol homeostasis, although it is most important in cells like macrophages that take up large amounts of exogenous cholesterol by various scavenger receptors, for example, a scavenger receptor class B type 1 (SR-B1) and by the phagocytosis of cholesterol-rich membranes from other cell types.
HDL, which contain apoA-I as their main protein, mediate the removal of excess cholesterol from peripheral cells and return it to the liver, where it can be directly excreted into the bile or converted into a bile salt. Cholesterol is also thought to be directly excreted by the intestine by the transintestinal cholesterol efflux pathway, but the details of this pathway are not fully understood. Because most cells besides hepatocytes cannot catabolize cholesterol, the reverse cholesterol transport pathway has been proposed to be their main atheroprotective function. HDL can be formed as a consequence of lipolysis of VLDL and CM, which causes the release of apoA-I and surface lipids that can then spontaneously reorganize as HDL particles. Most HDL, however, are probably formed by the liver and intestine when the ATP-binding cassette transporter A1 (ABCA1) on the plasma membranes adds phospholipids to newly secreted apoA-I, which contains a tandem array of amphipathic helices and acts like a protein detergent. In other tissues, ABCA1 also promotes the removal of excess cholesterol from peripheral cells to HDL and in this way helps cells maintain cholesterol homeostasis. Lecithin cholesterol acyltransferase (LCAT) is a key enzyme in the reverse cholesterol transport pathway, because it converts cholesterol to cholesteryl esters, thus trapping it in the core of HDL until it can be transported to the liver and taken up by the hepatic SR-B1. HDL, however, have many other potentially beneficial antiatherogenic properties, which are not fully understood, such as their antioxidant and antiinflammatory properties, that probably relate to their other protein and lipid components . Finally, CETP interconnects the reverse cholesterol transport pathway with the exogenous and endogenous pathways by exchanging cholesteryl esters on HDL with TG from apoB-containing lipoproteins.
Classification of dyslipidemias
Lipoprotein disorders can be classified as either being primary due to a genetic etiology ( Table 9.2 ) or secondary due to a metabolic or nutritional disorder or sometimes can be classified as iatrogenic as a consequence of a drug treatment ( Table 9.3 ). A common practical approach for classifying the various causes for dyslipidemia is to first determine which of the major lipids or lipoproteins are present in abnormal amounts, either high or low. Thus the various dyslipidemias will be discussed in this chapter under the headings hypercholesterolemia, hypertriglyceridemia, combined hyperlipidemia (high TG and total cholesterol, TC), hypolipidemia with hypoalphalipoproteinemia (low HDL-cholesterol, HDL-C), and hypotriglyceridemia. In addition, the original phenotypic classification by Fredrickson, D.S., which was based on agarose gel electrophoretic separation of lipoproteins , is also shown in Table 9.2
| Genetic disorder | Phenotype | Biochemical defect | Clinical presentation | TC | TG |
|---|---|---|---|---|---|
| Familial LPL deficiency | I | Absence of LPL activity | Eruptive xanthoma; hepatosplenomegaly; pancreatitis | Increased + | Increased +++ |
| Familial apoC-II deficiency | I or V | Absence or abnormal structure of apoC-II | Pancreatitis | Increased + | Increased +++ |
| Familial hypercholesterolemia | IIa or IIb | Deficiency of LDL receptors | Tendinous and tuberous xanthoma; premature atherosclerosis | Increased +++ | Normal (IIa) or increased + (IIb) |
| Familial dysbetalipoproteinemia | III | Abnormal apoE and defect in TG-rich lipoprotein metabolism | Tuberoeruptive and planar xanthoma; premature atherosclerosis | Increased ++ | Increased ++ |
| Familial combined hyperlipidemia | IIa, IIb, or IV | Increased apoB production | Premature atherosclerosis | Increased or normal | Increased or normal |
| Familial | IV and V | Unknown | Type V: eruptive xanthoma; hepatosplenomegaly; pancreatitis | Normal (IV) | Increased+ (IV) |
| hypertriglyceridemias | Increased + (V) | Increased +++ (V) |
| Predominantly hypercholesterolemia |
| Hypothyroidism |
| Cholestasis |
| Nephrotic syndrome |
| Dysglobulinemias |
| Diuretics |
| Cyclosporine |
| Predominantly hypertriglyceridemia |
| Obesity |
| Diabetes mellitus |
| Alcoholism |
| Renal failure |
| Lipodystrophy |
| Dysglobulinemias |
| Estrogen therapy |
| Steroid therapy |
| Beta-blocker therapy |
| Isotretinoin therapy |
| Hypoalphalipoproteinemia |
| Obesity |
| Diabetes |
| Physical inactivity |
| Smoking |
| High-carbohydrate diet |
| Anabolic steroids |
| Beta-blockers |
| Liver disease |
Hypercholesterolemia
Because much of the risk of high cholesterol is when it is in LDL, LDL-C is usually the main cholesterol parameter to classify patients at risk for CHD. According to the 2018 American Heart Association/American Association of Cardiology (AHA/ACC) guidelines an optimal LDL-C in adults is <100 mg/dL, near optimum 100–129 mg/dL, borderline high is 130–159 mg/dL, high 160–189 mg/dL, and very high is ≥190 mg/dL . Based on a wide variety of experimental and clinical studies, LDL-C is known to be causally related to the development of CHD. Furthermore, numerous studies have shown that lowering LDL-C, either by diet or in combination with a drug therapy, can reduce cardiovascular morbidity and mortality and even total mortality .
Having established that a patient has hypercholesterolemia with an elevated concentration of LDL-C, it is first important to exclude any secondary cause of hypercholesterolemia ( Table 9.3 ), which is best treated by addressing any underlying disorder. For example, hypothyroidism can present with elevated LDL-C and TG. It is more effective to treat it with thyroid hormone replacement therapy rather than a lipid-lowering drug, which will also address the other clinical consequences of the thyroid disease.
A prototypical genetic disorder that results in hypercholesterolemia is familial hypercholesterolemia, which is fairly frequent, with an incidence between 1/200 and 1/300 in the heterozygous state . This is typically an autosomal dominant disorder characterized by increased levels of LDL-C, usually due to a genetic defect in the LDLR. Patients that are homozygous for defects in the LDLR typically present in pediatric practice, and these children have few or no functional LDLRs, resulting in marked elevations in LDL-C, xanthoma formation on the skin and tendons, premature atherosclerosis, aortic valve disease, and myocardial infarction as early as the second decade of life. The more common variety, which presents in adults, is the heterozygote in whom there is about a 50% reduction in LDLR activity, resulting in about a twofold increase in LDL-C. They can also present with tendon xanthomas and premature atherosclerosis, usually manifesting in early to middle adulthood.
To date, numerous mutations of the LDLR gene have been described that affect either its synthesis, correct orientation on the cell, or its internalization by endocytosis. It has been shown that approximately 18% carriers and noncarriers of a mutation for LDLR are being misdiagnosed when using only cholesterol levels for their diagnosis .
Recently, defects in two auxiliary proteins important in the function of the LDLR, namely LDL receptor adaptor protein 1 (LDLRAP1) and proprotein convertase subtilisin/kexin type 9 (PCSK9), have also been described as a rare cause of familial hypercholesterolemia . LDLRAP1 is an adaptor protein important in the endocytosis of the LDLR and mutations in it are only apparent when both alleles are mutated, and thus it is an autosomal recessive form of hypercholesterolemia. PCSK9 is a secretory protein produced mainly by the liver that promotes the internalization and degradation of the LDLR. Loss-of-function mutations in PCSK9 are associated with low LDL-C and patients have reduced CHD risk, whereas patients with gain-of-function mutations have high LDL-C and have increased CHD risk. Recently a new therapy based on monoclonal antibodies against PCSK9 has been developed for markedly lowering LDL-C when given in conjunction with a statin .
Mutations in the receptor-binding region of apoB-100, at position 3500 and rarely elsewhere on apoB-100, have also been shown to result in a defective apoB-100 with decreased affinity to the LDLR, resulting in an elevated LDL-C. Clinically, this type of dyslipidemia is usually not as severe as familial hypercholesterolemia and has been named familial defective apoB. Familial hypercholesterolemia can also be subclassified into two types, depending on plasma TG. In the type IIa disorder, plasma TG are normal, whereas in the type IIb disorder, plasma TG are increased due to elevated levels of VLDL ( Table 9.2 ) and these patients may be at even more risk for CHD than type IIa.
Patients with elevated lipoprotein(a), Lp(a), can also present with hypercholesterolemia . Lp(a) is like LDL in structure but has an additional highly polymorphic protein that differs in size called apo(a), which is covalently attached to apoB-100 by a disulfide bond. Apo(a) resembles plasminogen in structure and may interfere with clot lysis, which is possibly the reason that Lp(a) is so proatherogenic. In fact, polymorphisms that affect its synthesis and the size polymorphisms of apo(a) are one of the strongest genetic risk factors for not only CHD but also aortic valve calcification. It may also be a cause for thrombotic stroke. There are currently no effective therapies for lowering Lp(a), but individuals who are at risk because of high levels of Lp(a) should be aggressively treated for other risk factors. About quarter of the population have elevated Lp(a) levels that put them at risk for CHD and not all these patients will have hypercholesterolemia, so patients that have a strong family history of CHD and/or do not respond well to statin therapy should ideally be tested by an immunoassay for Lp(a) .
Most patients who present with hypercholesterolemia have only modest increases in LDL-C when compared with familial hypercholesterolemia. In these cases, it is likely a consequence of a complex interaction of common genetic polymorphisms that affect lipid metabolism and environmental factors and has been referred to as polygenetic hypercholesterolemia. In contrast to familial hypercholesterolemia, these patients often show a good response to drug therapy and are also more likely to respond to lifestyle changes and thus have a better prognosis.
According to AHA/ACC 2018 guidelines , it is recommended that all adults 20 years or older be screened with a lipid panel (TC, TG, HDL-C, and a calculated or measured LDL-C) every 5 years. Treatment goals and drug approaches are primarily based on LDL-C levels and vary depending on the risk for CHD ( Fig. 9.2 ). Risk is determined for age 40–75 years, using a calculation based on the presence of clinically existing CHD or risk equivalents (diabetes mellitus, chronic kidney disease); on laboratory tests, such as LDL-C and TG concentrations ( Table 9.5 ); and risk-enhancing factors for CHD ( Table 9.4 ). Those patients at greatest risk should be treated more aggressively, which usually involves lipid-lowering drug treatment, most often a statin ( Fig. 9.2 ). Statins by inhibiting 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, the rate-limiting step in cholesterol biosynthesis, decrease the regulatory pools of cholesterol in hepatocytes. Statins then cause an upregulation of the LDLR and the lowering of plasma LDL. In contrast to previous recommendations, a nonfasting sample can be used as a screening test. If nonfasting TG is greater than 400 mg/dL, a fasting sample should be tested. Besides a lipid panel, for those patients at intermediate risk other laboratory tests, which are referred to as risk enhancers ( Table 9.4 ), can be used to decide therapy, such as apoB, Lp(a), and high-sensitivity C-reactive protein (hsCRP), a marker of inflammation. For those at the highest intermediate risk, cardiovascular imaging for coronary vessel calcification may also be helpful. For those age 20–39 years, they should be counseled to follow a healthy lifestyle and statins can be considered if there is a strong family history and LDL-C is greater than 160 mg/dL.