Abstract
In the last two decades, great progress has been made in refining the classification and elucidating the molecular basis of genetic lipodystrophies, rare disorders characterized by selective loss of body fat and predisposition to insulin resistance and its metabolic complications such as diabetes mellitus, hypertriglyceridemia, and hepatic steatosis. The autosomal recessive, congenital generalized lipodystrophy is due to mutations in AGPAT2 , BSCL2 , CAV1 , and PTRF ; and autosomal dominant familial partial lipodystrophy (FPLD) is due to mutations in LMNA , PPARG , AKT2 , and PLIN1 . Among extremely rare syndromes, autosomal recessive, mandibuloacral dysplasia is due to LMNA and ZMPSTE24 mutations and an autoinflammatory lipodystrophy syndrome due to PSMB8 mutations. SHORT syndrome is due to mutations in PIK3R1 and m andibular hypoplasia, d eafness, p rogeroid features syndrome due to POLD1 mutation. The precise mode of inheritance and molecular genetic bases of many extremely rare forms of genetic lipodystrophies remain to be elucidated.
Keywords
lipodystrophy, congenital generalized lipodystrophy, familial partial lipodystrophy, mandibuloacral dysplasia, lamin A/C, adipose tissue, autoinflammatory syndromes, AGPAT2, BSCL2, caveolin, PPARG, PTRF
Introduction
During the last two decades, major advances have been made in elucidation of the molecular genetic basis of inherited lipodystrophy syndromes. The first step toward identification of the causal genes required careful phenotyping of the patients based on clinical features and in-depth characterization of the body fat distribution using conventional anthropometry and whole body magnetic resonance imaging, followed by proper classification of the various types and subtypes of lipodystrophies. Initially, discoveries of the causal genes were made using the classical linkage analysis approach followed by positional cloning. More recently, elucidation of the molecular genetic basis of some extremely rare syndromes has been made possible, due to availability and utilization of the next generation, whole exome sequencing. This knowledge of the underlying basis of genetic lipodystrophies has further improved categorization of the lipodystrophies and refinement of the clinical features associated with various subtypes.
The lipodystrophies are characterized by selective loss of body fat ( Fig. 23.1 ) and a predisposition to developing insulin resistance and its complications, such as diabetes mellitus, hypertriglyceridemia, hepatic steatosis, polycystic ovarian syndrome, acanthosis nigricans, and hypertension. The loss of fat can be generalized (involving all the body fat depots), partial (affecting the limbs) or focal or localized (from discrete areas of the body). The severity of the metabolic complications is generally related to the extent of body fat loss. For example, patients with generalized lipodystrophies have more severe diabetes, hypertriglyceridemia, and hepatic steatosis than those with partial lipodystrophies.
The diagnosis of the various syndromes of lipodystrophies is mainly based on clinical findings, that is, history, physical examination, and anthropometry. The clinical diagnosis can be confirmed by genetic screening for those subtypes for which the causal genes are known. However, for many syndromes, the molecular genetic basis remains unknown, and therefore at present, only clinical diagnostic criteria can be applied to achieve a diagnosis. Genetic lipodystrophies can be broadly classified based on the pattern of inheritance, that is, autosomal recessive or autosomal dominant ( Table 23.1 ). The overall diagnostic approach to genetic lipodystrophies is presented in this chapter.
|
Autosomal recessive lipodystrophies
Congenital Generalized Lipodystrophy (CGL)
This syndrome was first reported in 1954 in two young Brazilian boys who were two and six years old and presented with marked hepatosplenomegaly, acromegaloid gigantism, fatty liver, and hyperlipidemia. Five years later, three additional patients from Norway were reported with generalized lack of body fat from birth and the disorder was termed congenital generalized lipodystrophy (CGL). However, many subsequent investigators have also used the terminology Berardinelli–Seip congenital lipodystrophy in recognition of the physicians who initially reported this syndrome. Because of striking clinical appearance at birth, the diagnosis of CGL is usually made by neonatologists or pediatricians. Only a few patients may have a normal appearance at birth, and loss of body fat may become evident later during the first year of life.
The precise population prevalence of CGL is not known. In the literature to date, ∼300 patients with CGL have been reported. Some regions from Brazil and from Lebanon have reported a cluster of cases, likely due to in-breeding. If it is assumed that only a quarter of the actual number of cases may be reported in the literature, the estimated worldwide prevalence of CGL may be about one in 10 million. Recent establishment of worldwide and regional patient registries may help in better estimates of prevalence of CGL and other genetic lipodystrophies in future. Further refinement of the diagnostic codes in the ICD-10 system may also be advantageous in estimating the prevalence of genetic lipodystrophies.
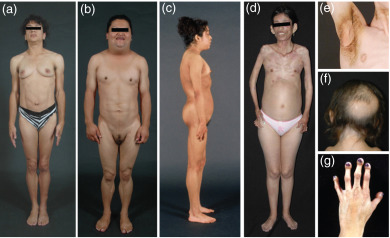
Patients with CGL present with muscular appearance at birth instead of the normal chubby appearance of the newborns. This is due to near complete absence of body fat ( Fig. 23.1 a). The face may look gaunt. It is likely that the parents and caregivers may not recognize this abnormal phenotype in male newborns, but will be able to recognize it in female infants. The affected children demonstrate an accelerated growth with an insatiable appetite. They have acromegaloid features with enlarged mandible, hands, and feet. Prominence of umbilicus or umbilical hernias are noticed at birth likely due to underlying hepatomegaly and/or splenomegaly. Patients with CGL do not have acanthosis nigricans at birth; however, it appears during early childhood or after puberty, and can affect many body regions including the typical neck, axillae, and groin regions, and other regions such as the trunk, hands, knees, elbows, and ankles ( Fig. 23.1 e).
Liver enlargement due to hepatic steatosis is usually noticed during infancy. A few patients develop steatohepatitis followed by fibrosis. Some do develop cirrhosis or end-stage liver disease and its complications later on in life requiring hepatic transplantation. In females, mild hirsutism, clitoromegaly, and irregular menstrual periods are common and some present with primary or secondary amenorrhea and polycystic ovaries. In some women, clitoromegaly can occur without any signs of hirsutism. Breast development is normal but the overlying subcutaneous fat layer surrounding the mammary tissue is absent. Most affected women are unable to conceive; however, a few patients have had successful pregnancies. The pregnancy in these patients is high risk and may require management of diabetes as well as severe hypertriglyceridemia. Affected men usually have normal reproductive ability.
In some patients, radiographs reveal focal lytic lesions in the long bones such as the humerus, femur, radius, ulna, carpal, tarsal, or phalangeal bones after puberty, which may be confused with polyostotic fibrous dysplasia. The lesions may develop due to lack of ability to make normal bone marrow fat to replace hematopoietic marrow during childhood and adolescence. Instead, the marrow may be replaced by vascular tissue. The pathological fractures in the affected bones are likely and children with these lesions may be advised to avoid contact sports. Some patients have also been reported to have hypertrophic cardiomyopathy and mild mental retardation.
Even during infancy and early childhood, patients with CGL develop extreme hyperinsulinemia, and a few will also develop hyperglycemia. However, most of the patients develop diabetes during the pubertal years. An autopsy study revealed marked amyloidosis of pancreatic islets affecting more than 90% of the islets and β cell atrophy in a young adult female with CGL. Thus, extreme insulin resistance from birth may induce premature and severe amyloidosis and β cell death, similar to that observed in patients with type 2 diabetes mellitus. Diabetes, however, is extremely challenging to manage in these patients and frequently requires extremely high doses of insulin for glycemic control. Interestingly, diabetes is ketosis resistant likely due to endogenous hyperinsulinemia.
Most patients with CGL develop hypertriglyceridemia later during childhood and adolescence; however, some develop extreme hypertriglyceridemia during infancy. This type 5 hyperlipoproteinemia with chylomicronemia predisposes many patients to recurrent episodes of acute pancreatitis particularly those with uncontrolled diabetes. The levels of high density lipoprotein (HDL) cholesterol also tend to be low. However, atherosclerotic vascular complications such as coronary heart disease, strokes, or peripheral vascular disease have not been reported in the literature. On the other hand, most of the reported subjects have been children. As we follow more patients with CGL into late adulthood, we will be able to ascertain cardiovascular implications of type 5 hyperlipoproteinemia in them.
Because of near total loss of fat in patients with CGL, the levels of serum leptin and adiponectin are markedly reduced. However, extremely low levels of serum leptin should not be considered diagnostic. Serum leptin assays are not standardized and marked variability is observed in levels measured by various commercial kits. Severe hypoleptinemia may induce voracious appetite in patients with CGL, which may further contribute to metabolic complications.
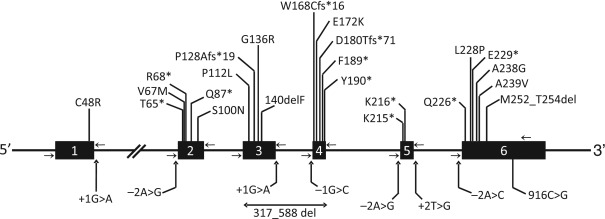
To date, four distinct genetic subtypes of CGL have been reported. Type 1 CGL due to mutations in the 1-acylglycerol-3-phosphate- O -acyltransferase 2 ( AGPAT2 ) gene and type 2 due to mutations in the Berardinelli–Seip congenital lipodystrophy 2 ( BSCL2 ) gene are the most common subtypes. Type 3 CGL due to a homozygous nonsense mutation in caveolin 1 ( CAV1 ) was reported in a single patient and type 4 CGL due to mutations in polymerase I and transcript release factor ( PTRF ) has been reported in about 30 patients.
CGL Type 1: AGPAT2 Mutations
These patients have a characteristic body fat distribution on whole body magnetic resonance imaging. They have near total absence of adipose tissue from most subcutaneous areas, intra-abdominal and intrathoracic regions and bone marrow (metabolically active) but adipose tissue in the palms, soles, under the scalp, orbital, and periarticular regions is spared (mechanical adipose tissue). They also show increased predisposition to develop lytic bone lesions; mostly affecting long bones of the extremities.
The AGPAT enzymes play an essential role in the biosynthesis of triglycerides and phospholipids. Currently, 11 isoforms of AGPAT, each encoded by a different gene, are known. These enzymes add an acyl (fatty acid moiety) to lysophosphatidic acid (1-acylglycerol-3-phosphate) to form phosphatidic acid (1,2-diacylglycerol-3-phosphate). Phosphatidic acid can be further acylated and processed to form triglycerides or other phospholipids. Each isoform has a unique tissue expression and the AGPAT2 isoform is highly expressed in the adipose tissue. Thus, deficiency of AGPAT2 may impair triglyceride and phospholipid biosynthesis in the adipocytes and result in lipodystrophy.
Most of the patients harbor null mutations with no enzymatic activity demonstrable in vitro ( Fig. 23.2 ). However, some compound heterozygotes have a null and a missense mutation (with some residual enzymatic activity) and a few have homozygous missense mutations. However, so far the type of mutation does not seem to determine the phenotype or the loss of fat and all the patients lack nearly all body fat. Nearly all patients of African origin harbor the founder mutation, c.IVS4-2A>G (p.Gln196fsX228), on one or both alleles.
CGL Type 2: BSCL2 Mutations
As compared to patients with type 1 CGL, patients with CGL type 2 have an increased prevalence of cardiomyopathy and mild mental retardation. They have almost total lack of body fat with both metabolically active and mechanical adipose tissue being absent. As compared to patients with type 1 CGL, their serum leptin levels are lower, but serum adiponectin levels are higher. Recently, teratozoospermia was reported in a CGL type 2 patient with sperm defects including abnormal head morphology, bundled sperm with two or more sperm connected to each other with large ectopic lipid droplets.
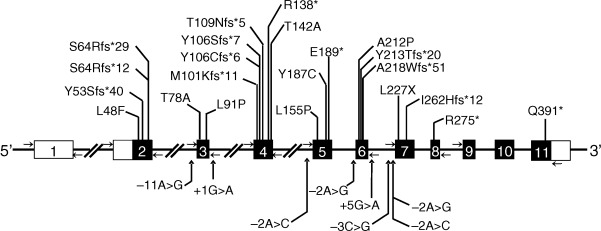
The BSCL2 encodes a 398 amino acid transmembrane protein called seipin with no significant homology to any other known protein. It has a CAAX motif at the carboxy-terminus and a glycosylation site, NVS, at position 88-90. Recent data suggest the role of seipin in lipid droplet formation and in adipocyte differentiation. Studies with the seipin homolog in the yeast suggest that it may be playing a role in fusion of lipid droplets. Seipin has been shown to bind phosphatidic acid phosphatase (lipin 1) and thus may also play a role in phospholipids and triglyceride synthesis. Nearly all BSCL2 mutations reported in patients with CGL2 are null ( Fig. 23.3 ). However, a few have been missense mutations. So far, no phenotypic differences have been reported between those with null and missense mutations.
CGL Type 3: Caveolin 1 Mutations
A 20-year-old Brazilian girl with generalized lipodystrophy, short stature, and presumed vitamin D resistance has been reported to have a homozygous CAV1 mutation. She had hepatic steatosis and splenomegaly and developed diabetes at 13 years of age. Other clinical features included acanthosis nigricans, severe hypertriglyceridemia, primary amenorrhea, and functional megaesophagus. Mechanical adipose tissue and fat in the bone marrow was not affected by lipodystrophy.
Caveolin 1 is a major component of caveolae. Caveolae are specialized plasma membrane microdomains appearing as 50–100 nm vesicular invaginations. CAV1 is expressed ubiquitously but is highly expressed in the adipocytes, endothelial cells, and fibroblasts. Caveolins play an important role in vesicular trafficking, homeostasis of cellular cholesterol, fatty acids and triglycerides, and signal transduction. Caveolae may contribute lipids to the lipid droplets and CAV1 mutation may cause lipodystrophy due to defective lipid droplet formation.
CGL Type 4: PTRF Mutations
About 30 patients have been reported to have CGL type 4 due to PTRF . Besides having generalized lipodystrophy, they have congenital myopathy with high serum creatine kinase levels. Other clinical features include pyloric stenosis, atlantoaxial instability, and predisposition to serious arrhythmias such as catecholaminergic polymorphic ventricular tachycardia, prolonged QT interval, and sudden death. Mechanical and bone marrow fat is well preserved. These patients may not show severe lipodystrophy at birth and may lose body fat progressively during infancy. PTRF regulates the expression of caveolins 1 and 3 and thus plays an important role in biogenesis of caveolae.
Molecular Diagnosis
The first step is to achieve a clinical diagnosis based on history and physical examination. Clinically the major differentiating clinical finding between the two major types of CGL, type 1 and 2, is the presence or absence of subcutaneous fat from the palms and soles, respectively. CGL type 4 can be easily distinguished from the other subtypes by the presence of myopathy and high serum creatine kinase levels. Molecular diagnosis at research laboratories is available for confirmation. Molecular diagnosis may be helpful for understanding the risk of having another child with CGL and can also be used for prenatal screening. Besides patients with known genotypes, there are a few patients with CGL who do not have mutations in any of the four known loci and thus there may be novel genes to be discovered for CGL.
Differential Diagnosis
CGL should be differentiated from acquired generalized lipodystrophy, leprechaunism, atypical progeroid syndrome, generalized lipodystrophy due to LMNA mutations, and neonatal progeroid syndrome. Patients with acquired generalized lipodystrophy develop fat loss after birth and are likely to have associated autoimmune diseases or panniculitis. Patients with generalized lipodystrophy or atypical progeroid syndrome due to LMNA mutations have normal body fat at birth and lose it later in childhood.
Mandibuloacral Dysplasia (MAD)-Associated Lipodystrophy
Mandibuloacral dysplasia is an extremely rare autosomal recessive disorder, which has been reported in about 40 patients so far. Clinical features include mandibular hypoplasia, resorption of lateral parts of the clavicles and acro-osteolysis (resorption of the terminal phalanges) ( Fig. 23.1 c,f,g). Patients may also present with delayed closure of cranial sutures, joint contractures, mottled cutaneous pigmentation, short stature, and other skeletal deformities. Patients with MAD also develop “progeroid features” such as the bird-like facies, high-pitched voice, skin atrophy, pigmentation, alopecia, and nail dysplasia. They develop either partial lipodystrophy affecting the extremities (type A) or more generalized lipodystrophy, which affects the face, trunk, and extremities (type B). Metabolic complications such as hyperinsulinemia, insulin resistance, impaired glucose tolerance, diabetes mellitus, and hyperlipidemia are usually mild to moderate in severity.
MAD Type A Due to LMNA Mutations
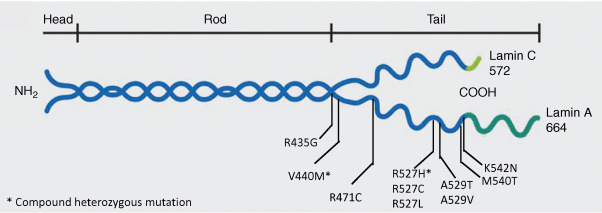
Novelli et al. reported a homozygous missense p.Arg527His mutation in the lamin A/C ( LMNA ) gene in MAD patients with type A (partial) lipodystrophy of Italian origin, which appears to be a founder mutation. LMNA has 12 exons and by alternative splicing in exon 10, it encodes for either a short protein, lamin C, or a full-length protein, prelamin A. While lamin C does not undergo posttranslational processing, prelamin A undergoes a series of modifications to form the mature protein called lamin A. This processing occurs at the extreme C-terminal of the prelamin A, which has a C-A-A-X motif. The first step in posttranslational processing of prelamin A involves farnesylation of cysteine residue in the C-A-A-X motif by a farnesyl transferase, which is followed by proteolysis of the tripeptide, A-A-X, by the enzyme zinc metalloprotease (ZMPSTE24). Then, the cysteine residue is carboxymethylated and finally ZMPSTE24 or another protease cleaves another 15 residues from the C-terminus forming mature lamin A. Lamins A and C, encoded by LMNA , and lamins B1 and B2, encoded by other genes, belong to the intermediate filament family of proteins and from heterodimeric or homodimeric coiled-coil structures, which constitute nuclear lamina, located between the inner nuclear membrane and chromatin. Lamins provide structural integrity to the nuclear envelope and interact with chromatin and several other nuclear envelope proteins. So far, a total of 30 patients with MAD due to various LMNA mutations have been reported ( Fig. 23.4 ). Some patients have severe clinical manifestations including alopecia, loss of eyebrows, delayed sexual maturation, premature loss of teeth, as well as early death. Most of the LMNA mutations causing MAD are located in the C-terminal region affecting exons 8–10 with p.R527H being the most common ( Fig. 23.4 ). How these specific LMNA mutations cause resorption of bones such as the mandible, clavicles, and terminal phalanges remains unclear. Skin fibroblasts from some of the patients show nuclear morphological abnormalities such as budding, invaginations of the nuclear membrane, similar to those seen in patients with Hutchinson–Gilford progeria syndrome. Whether there is accumulation of prelamin A in skin fibroblasts from these patients is controversial.
MAD, Type B Associated with Zinc Metallopeptidase STE24 (ZMPSTE24) Mutations
Recognizing the critical role of ZMPSTE24 in prelamin A processing, our group considered ZMPSTE24 as a candidate gene for MAD and reported compound heterozygous mutations in ZMPSTE24 in a Belgian woman with MAD, progeroid features, and generalized lipodystrophy. She died prematurely at age 24 years due to end-stage renal disease caused by focal segmental glomerulosclerosis. It is suggested that accumulation of prelamin A and/or lack of mature lamin A in the cells may be the underlying mechanism of cellular toxicity in these patients. A total of eight patients with this subtype have been reported and most of them have been young children. There are no reports of diabetes among them. As compared to MAD patients with LMNA mutations, those with ZMPSTE24 mutations are premature at birth, have early-onset of skeletal defects including acro-osteolysis, have more severe progeroid appearance, and develop subcutaneous calcified nodules on the phalanges. They are also prone to develop renal disease.
Molecular and Differential Diagnosis
Clinical diagnosis should be established first followed by molecular genetic diagnosis. Genotyping may help in predicting clinical course and complications in the patients. Differential diagnosis should include Hutchinson–Gilford progeria syndrome, and atypical progeroid syndrome (mostly due to heterozygous missense mutations in LMNA ), and other disorders presenting with acro-osteolysis including Hajdu–Cheney ( NOTCH2 mutations), Haim–Munk, and Papillon–Lefevre syndromes (both due to Cathepsin C mutations).
Mandibular Hypoplasia, Deafness, Progeroid Features (MDP)-Associated Lipodystrophy Syndrome
This syndrome was reported by our group based on their unique clinical features including m andibular hypoplasia, d eafness, p rogeroid features (MDP)-associated lipodystrophy. They showed distinct characteristics such as sensorineural hearing loss, and absence of clavicular hypoplasia and acro-osteolysis. All males with MDP had undescended testes and hypogonadism. One adult female showed lack of breast development. Two of the seven patients had diabetes mellitus. Recently, a recurrent de novo in-frame deletion (c.1812_1814delCTC, p.Ser605del) of a single codon in polymerase (DNA- directed) delta 1 catalytic subunit ( POLD1 ) was reported in patients with MDP syndrome. DNA polymerase δ cooperates with WRN, a DNA helicase, and serves an important role in maintaining genome stability.
Autoinflammatory Lipodystrophy Syndrome
Recently, we reported in depth a phenotype of an autosomal recessive, autoinflammatory, j oint contractures, m uscle atrophy, m icrocytic anemia, and p anniculitis-induced lipodystrophy (JMP) syndrome in two pedigrees. Patients with JMP syndrome had onset of progressive panniculitis-induced lipodystrophy during childhood. Histopathology of the subcutaneous fat upon biopsy reveals infiltration of the adipose tissue with lymphocytes, neutrophils, and multinucleated giant cells called panniculitis. When these lesions with panniculitis heal, they result in localized loss of subcutaneous fat from a small area but with time it can affect the entire region such as the face, neck, upper trunk, and arms and legs ( Fig. 23.1 d). Previously, some patients with a similar but milder clinical phenotype were reported from Japan. These patients also have overlapping features with recently reported patients with chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. All these patients with autoinflammatory lipodystrophy have variable clinical features including intermittent fever, hypergammaglobulinemia, elevated erythrocyte sedimentation rate, hepatosplenomegaly, and calcification of basal ganglia. A total of 20 patients (2–51 years old) have been reported so far.
We initially reported a homozygous, missense, loss of function, c.224C>T (p.Thr75Met) mutation in proteasome subunit, beta-type, 8 ( PSMB8 ) gene in patients with JMP syndrome and subsequently the patients from Japan as well as those with CANDLE syndrome were reported also to have mutations in PSMB8 . PSMB8 encodes the β5i subunit of the immunoproteasome. Immunoproteasomes play a critical role in proteolysis of antigens presented by major histocompatibility complex class I molecules and result in generation of immunogenic epitopes. The mutations in PSMB8 may trigger an autoinflammatory response resulting in panniculitis and other clinical manifestations.
S hort Stature, H yperextensibility of Joints and/or Inguinal H ernia, O cular Depression, R eiger Anomaly, and T eething Delay (SHORT) Syndrome
A total of 30 patients have been reported with SHORT syndrome. The pedigrees reveal both autosomal recessive and dominant modes of transmission. Reiger anomaly consists of eye abnormalities such as iris hypoplasia, Schwalbe ring, iridocorneal synechiae, micro- or megalocornea, and dental anomalies such as hypodontia, microdontia, enamel hypoplasia, and atypical teeth. Other clinical features include intrauterine growth retardation, failure to thrive, delayed speech development, small head circumference, bilateral clinodactyly, and sensorineural hearing loss. Different patterns of fat loss have been reported. In many patients, lipodystrophy affects the face, upper extremities, and sometimes the trunk, with relative sparing of the lower extremities. Others have lipodystrophy affecting only the face, gluteal region, and elbows. Diabetes occurs as early as the second and third decade of life. The pathogenesis of diabetes mellitus remains unclear. Recently, patients with SHORT syndrome were reported to harbor de novo or autosomal dominantly inherited heterozygous mutations in phosphatidylinositol 3-kinase regulatory subunit 1 ( PIK3R1 ). PIK3R1 encodes multiple regulatory subunits of phosphatidylinositol 3-kinase (PI3K), an intracellular enzyme with a central role in insulin signaling. The molecular genetic basis of the autosomal recessive variety of SHORT syndrome remains to be determined.
Neonatal Progeroid Syndrome (Wiedemann–Rautenstrauch Syndrome)
This syndrome has been described in about 25 cases. The clinical features are evident at birth but are heterogeneous. Patients have a triangular, old-looking face with relatively large skull (progeroid appearance), prominent veins on the scalp, sparse scalp hair, large anterior fontanelle, and generalized lipodystrophy. However, subcutaneous fat in the sacral and gluteal areas is spared. Approximately half of the patients reportedly died before the age of 6 years but some patients surviving up to the age of 16 years have been reported. Recently, a few patients with overlapping features of progeroid and Marfan’s syndromes were reported to harbor de novo heterozygous null mutations in the penultimate exon of fibrillin 1 ( FBN1 ) gene. These patients have been suggested to be having progeroid fibrillinopathy. Many others do not harbor FBN1 variants suggesting additional loci for this syndrome.
Familial Partial Lipodystrophy (FPLD) Due to CIDEC Mutation
A single patient has been reported to have autosomal recessive FPL FPLD, type 5 due to a homozygous missense mutation in cell death-inducing DNA fragmentation factor a-like effector c ( CIDEC ). This 19-year-old girl from Ecuador developed diabetic ketoacidosis at age 14 and also had hypertriglyceridemia and hypertension. Interestingly, subcutaneous fat biopsy revealed multilocular, small lipid droplets in adipocytes, which had been previously reported in the Cidec knockout mice.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


