Leukemia and ABC Transporters
Yu Fukuda; Shangli Lian; John D. Schuetz1 Department of Pharmaceutical Sciences, St. Jude Children’s Research Hospital, Memphis, Tennessee, USA
1 Corresponding author: email address: john.schuetz@stjude.org
Abstract
Acute myeloid leukemia (AML) is a heterogeneous disease caused by aberrant proliferation and/or differentiation of myeloid progenitors. However, only ~ 65% of AML patients respond to induction chemotherapy and the overall survival rate for AML remains low (~ 24% for 5-year survival). The conventional view suggests that ATP-binding cassette (ABC) transporters contribute to treatment failure due to their drug-effluxing capabilities. This might be overly simplistic. Some ABC transporters export endogenous substrates that have defined roles in normal hematopoietic progenitors. It is conceivable that these substances also provide an advantage to leukemic progenitors. This review will highlight how certain endogenous substrates impact normal hematopoietic cells and suggest that ABC transporters facilitate export of these substances to affect both normal hematopoietic and leukemic progenitors. For example, the ability to export certain endogenous ligands may facilitate leukemogenesis by modifying leukemic progenitor cell proliferation or survival. If so, the addition of ABC transporter inhibitors to traditional chemotherapy might improve therapeutic efficacy by not just increasing intracellular drug accumulation but also blocking the beneficial effects ABC transporter ligands have on cell survival.
Abbreviations
ABC ATP-binding cassette
ALA δ-aminolevulinic acid
AML acute myeloid leukemia
CK casein kinase
CML chronic myelogenous leukemia
CMP common myeloid progenitor
COX cyclooxygenase
GMP granulocyte-macrophage progenitor
GPCR G protein-coupled receptor
HIF hypoxia-inducible factor
HPC hematopoietic progenitor cell
HSC hematopoietic stem cell
IBD inflammatory bowel disease
LSC leukemic stem cell
LSK Lin−Sca-1+c-Kit+
LTC4 leukotriene C4
MDS myelodysplastic syndrome
MEP megakaryocyte/erythroid progenitor
MPP multipotent progenitors
MRP multidrug-related protein
PGE2 prostaglandin E2
PPIX protoporphyrin IX
SNP single nucleotide polymorphism
1 Hematopoiesis and Leukemia
1.1 Hematopoietic stem cells and ABC transporters
Hematopoietic stem cells (HSCs) have the potential to undergo self-renewal and differentiation into multiple lineage committed blood cells. HSCs are characterized by their ability to repopulate the bone marrow, and single HSC gives rise to all blood cell types in circulation (Osawa, Hanada, Hamada, & Nakauchi, 1996; Smith, Weissman, & Heimfeld, 1991). By transplanting genetically marked mouse bone marrow cells into lethally irradiated recipient mice, Till and McCulloch showed that each spleen colony-forming unit (CFU) originated from a single clonogenic cell (Becker, McCulloch, & Till, 1963). Subsequent studies have shown that HSCs and their progenitors can be identified and isolated using cell surface markers (Spangrude, Heimfeld, & Weissman, 1988). Long-term HSCs have an unlimited self-renewal capacity, whereas short-term HSCs have limited self-renewal ability. The HSC progeny gives rise to multipotent progenitors (MPPs), which then differentiate to lineage-restricted progenitors including the myeloid progenitors (Fig. 1). It is generally acknowledged that HSCs show properties such as quiescence, drug resistance (through the expression of several ATP-binding cassette (ABC) transporters), an active DNA-repair capacity, and resistance to apoptosis, which enable long life span. Murine HSCs are characterized by the absence of markers for committed blood lineage (Lin−) and expression of c-Kit and Sca-1 (Lin−Sca-1+c-Kit+, LSK). Additional cell surface markers can discriminate long- and short-term HSCs as well as MPPs. Common lymphoid progenitors (CLP) express IL-7R, whereas other committed progenitors do not. Oligopotent myeloid progenitors lose Sca-1 expression, and a combination of FcγR and CD34 expression identifies common myeloid progenitors (CMPs), granulocyte-macrophage progenitors (GMPs), and megakaryocyte/erythroid progenitors (MEPs). The hierarchy of myeloid progenitors was determined by a combination of morphology and surface markers that was identified in each progenitor population (Akashi, Traver, Miyamoto, & Weissman, 2000). For example, CMPs were capable of generating all myeloerythroid cells, whereas GMP generated only granulocyte/macrophage (GM) cells and MEP formed only megakaryocyte/erythroid cells in their colonies.
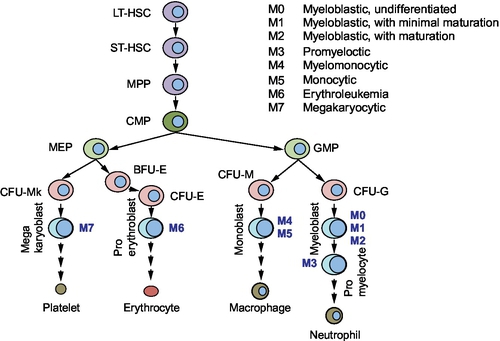
In humans, HSCs are identified by the cell surface markers, CD34 and CD38. Lineage-negative CD34+CD38− cells are enriched for progenitors capable of repopulating bone marrow of NOD–SCID mice with all lineages. Committed progenitors acquire the expression of CD38, and the human counterparts of CMP, GMP, and MEP were identified in the Lin−CD34+CD38+ population. Differentiation of progenitors into the myeloid lineage requires growth factors such as interleukin-3, GM-colony-stimulating factor, steel factor (c-Kit ligand), and erythropoietin as well as various transcription factors including runt-related transcription factor-1.
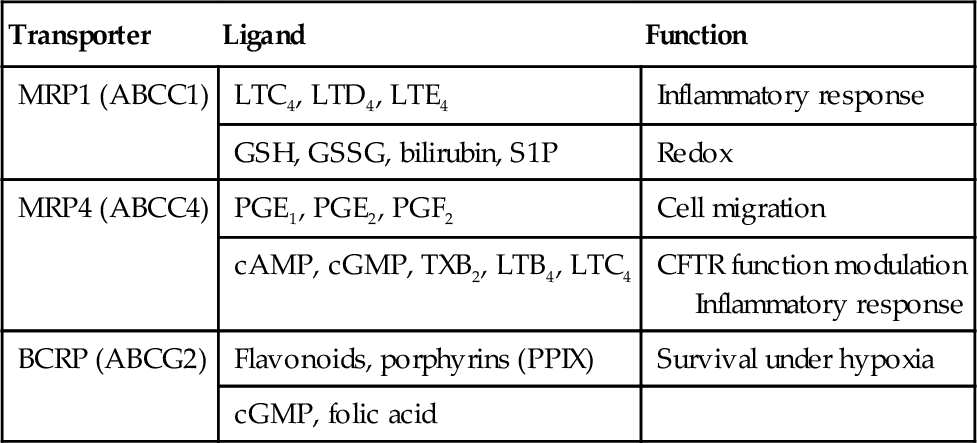
HSCs capacity to extrude toxins is likely related to high levels of ABC transporters that can protect cells from accumulation of endogenous and exogenous cytotoxic compounds (Table 1). In addition, the export of small molecules might serve a signaling function (e.g., leukotriene C4, LTC4). Side population (SP) cells are enriched for stem cells and are characterized by their ability to efflux the fluorescent dye, Hoechst 33342 (Goodell, Brose, Paradis, Conner, & Mulligan, 1996). ABCG2 was identified as the transporter responsible for Hoechst 33342 efflux which conferred the SP phenotype (Zhou et al., 2002). Absence of Abcg2 in hematopoietic cells did not affect normal unperturbed hematopoiesis. However, absence of Abcg2 rendered HSC markedly sensitive to chemotherapeutic agents that were Abcg2 substrates (e.g., mitoxantrone). Abcg2 may not protect all stem cells as embryonic stem cells or neural stem cells lack high Abcg2 expression (Ramalho-Santos, Yoon, Matsuzaki, Mulligan, & Melton, 2002).
1.2 Leukemic stem cells
Cancer stem cells are thought of as a rare group of cells that have self-renewal capacity coupled with the ability to produce tumors from a small number of initiating cells. The frequency of these cancer stem cells reportedly ranges from 0.0001% to 0.1% of the total cells; however, they may not be that rare (in some cases, e.g., myeloma; Huff & Matsui, 2008). Cancer stem cells were first identified in leukemia, but have also been identified in solid tumors such as brain, breast, cervix, and prostate (Dean, Fojo, & Bates, 2005). The leukemia-initiating cell population was identified and characterized 20 years ago (Bonnet & Dick, 1997; Lapidot et al., 1994). In acute myeloid leukemia (AML), the cell surface of leukemic stem cells (LSCs) resembles normal HSCs by both displaying the CD34+CD38− surface marker and possessing the ability to self-renew, proliferate, and differentiate. LSCs occur at an estimated frequency of 1 per 2 × 105 cells and produce AML when transplanted into NOD–SCID mice. In comparison, AML cells with the surface marker CD34+CD38+ have properties in common with more committed cells, i.e., limited self-renewal, and are nontumorigenic. These initial studies reported that regardless of the AML subtype (see Fig. 1), only CD34+CD38− AML cells produced leukemia in NOD–SCID recipient mice.
However, there are exceptions to the idea that only CD34+CD38− AML cells initiate leukemia. For instance, acute promyelocytic leukemia (French-American-British (FAB) class M3) is caused by an oncogenic PML–RARA fusion gene that is only detected in the CD34+CD38+ population. In this case, the CD34+CD38− cells were not tumorigenic. Moreover, recent studies using mouse leukemia models have suggested that the target cells for some oncogenes, such as MLL-AF9 and MOZ-TIF2, initiate AML in more committed progenitors such as the GMP (Huntly et al., 2004; Somervaille & Cleary, 2006). These LSCs resemble committed myeloid progenitors, but appear to have acquired the capacity for self-renewal, a property absent in their normal counterparts. Regardless of the origin, LSCs are self-renewing clonogenic cells that produce AML.
Self-renewal, increased proliferation, and altered differentiation are key features of leukemia cells. Notably, constitutive activation of pathways that drive proliferation is often observed in LSCs. For example, Wnt/β-catenin signaling is important in normal HSCs but is highly active in chronic myelogenous leukemia (CML) cells that are rapidly proliferating during blast crisis. PI3K activates signaling pathways such as Wnt and mTOR and is activated in AML. In contrast, PTEN, a PI3K antagonist that regulates self-renewal of normal HSC, is often downregulated in AML.
1.3 AML chemotherapy and ABC transporters
Chemotherapy is the mainstream treatment for AML; however, the development of drug resistance is a major obstacle in successfully achieving remission. As exporters of a variety of classes of chemotherapeutics, ABC transporters are considered to play a role in multidrug resistance in AML. For example, ABCB1/P-glycoprotein (P-gp) exports structurally diverse cancer chemotherapeutics including anthracyclines, one of the main chemotherapeutics used in the treatment of AML, and a marker for poor prognosis in AML (reviewed extensively in Shaffer et al., 2012). Because of its clinical relevance, P-gp inhibitors have been implemented in clinical trials to investigate if their addition to standard chemotherapeutic regimens improves clinical outcome. The results have been disappointing thus far and appear due to the nonspecificity of the inhibitors as well as the pharmacokinetic effects due to altered metabolism (Libby & Hromas, 2010). A widely accepted role for ABC transporters in AML chemotherapy is associated with their capacity to export drugs, thereby limiting leukemic cell drug exposure (Shaffer et al., 2012); however, other modes of resistance may involve intracellular drug sequestration.
Indeed, overexpression of ABCA3 increased lysosomal mass and lysosomal drug retention. This drug sequestration correlated with reduced cytotoxicity (Chapuy et al., 2009; Steinbach et al., 2006; Wulf et al., 2004). In this review, we focus on the potential role of some ABC transporters (in particular ABCC1, ABCC4, and ABCG2) in leukemia biology through their ability to modulate endogenous ligands.
2 ABC Transporters That Export Regulatory Molecules
2.1 Cyclic nucleotides—cAMP
The cyclic nucleotide, cyclic adenosine monophosphate (cAMP), is generated by adenylate cyclase in response to extracellular ligands and mediates a broad range of cellular responses through activation of protein kinase A. Intracellular levels of cAMP are modulated by factors such as phosphodiesterases and export (Cheepala et al., 2013). However, cyclic nucleotides are also compartmentalized into domains in the plasma membrane (Li et al., 2007; Sinha et al., 2013). Cyclic AMP is produced from ATP when adenylate cyclase is activated by G protein-coupled receptors (GPCRs). In the bone marrow niche, several GPCRs including β-adrenergic receptors, prostaglandin E2 (PGE2) receptors, and the chemokine MIP-1α receptors play an important role in affecting hematopoietic cell cAMP levels. Cyclic nucleotide-regulated pathways are involved in hematopoietic progenitor cell (HPC) proliferation and differentiation. The bone marrow is densely innervated by β-adrenergic fibers, and PGE2 and MIP-1α are secreted by monocytes and macrophages. Therefore, it is not surprising that HPCs contain high concentrations of cAMP-dependent PKA (Kobsar et al., 2008). Activation of PKA mediates a cascade of signaling events including PI3K/Akt and mTOR pathways.
Interestingly, cAMP inhibits proliferation of the myeloid leukemia cell lines, human Mo7e, and the murine myeloid progenitor cell line, 32D (Hendrie & Broxmeyer, 1994; Lee, 1999). Furthermore, the stable cAMP analog, 8-chloroadenosine 3′,5′-cyclic monophosphate (8-CL-cAMP), inhibited the self-renewal capacity of blast progenitors from acute myeloblastic leukemia patients (Pinto et al., 1992). Stimulation of PKA inhibits HPC proliferation in HPCs from tumor patients (Kobsar et al., 2008). Thus, it appears, in myeloid leukemia cells, that elevation of cAMP and PKA activation inhibits myeloid leukemia proliferation, unless factors counterbalance this.
2.2 MRP4 and cAMP
Within the C subfamily of ABC transporters, 13 full-length multidrug-related proteins (MRPs; i.e., encode two membrane-spanning domains and two nucleotide-binding domains in one transcript) have been identified, together with the cystic fibrosis transmembrane conductance regulator (ABCC7) and the sulfonylurea receptors (ABCC8 and ABCC9). MRP4/ABCC4 was the first ABC transporter shown to export nucleotide monophosphates (Schuetz et al., 1999), of which one of them (PMEA aka adefovir) resembled cAMP. Thus, it was not surprising that cAMP and cGMP were identified as endogenous substrates (Chen, Lee, & Kruh, 2001). Subsequent studies have identified other endogenous molecules such as ADP, eicosanoids, urate, bile acids, and conjugated steroid hormones as potential in vivo substrates (Ritter et al., 2005). Other ABCC family members MRP5 and 8 have also been shown to transport cAMP and cyclic guanosine monophosphate (cGMP), but these will not be discussed here as ABCC8 is discussed in the review by Nies et al. (Chapter 8).
MRP4 modulates the cAMP concentrations in cells in two ways: by affecting internal concentration by export and creating domains or gradients of cAMP concentration within the plasma membrane. In this context, MRP4 can reduce the local concentration of membrane by “flipping” cAMP from the membrane, to locally reduce its concentration (Li et al., 2007). MRP4 can also directly export cAMP as was originally shown by Kruh and colleagues (Chen et al., 2001). Distinguishing between MRP4 flippase and transport activity is not easily done.
Absence of MRP4 profoundly affects how cells migrate. For example, fibroblasts lacking MRP4 have both elevated membrane and intracellular cAMP and exhibit an enhanced rate of migration (Sinha et al., 2013). It is unknown if the migration of leukemic or hemapoietic cells is impacted in a similar way. Furthermore, how cAMP in the membrane versus intracellular cAMP affects proliferation and differentiation of normal hematopoietic and leukemia cells is unknown (Copsel et al., 2011).
2.3 Prostaglandins
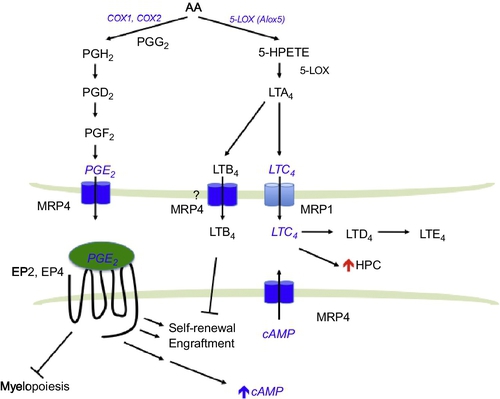
Prostaglandins are potent lipids derived from phospholipase-released arachidonic acid (AA) that are involved in numerous homeostatic biological functions and inflammation. Prostaglandins are synthesized de novo by prostaglandin H synthase (PGHS; referred to as COX for cyclooxygenase) when cells are activated by various external stimuli. PGHS exists as two isoforms referred to as PGHS-1 (COX-1) and PGHS-2 (COX-2; Fig. 2). In general, COX-1 is the enzyme responsible for basal, constitutive prostaglandins synthesis, whereas COX-2 is important in various inflammatory and “induced” settings. Prostaglandins are formed and “released” by most cells to act as autocrine- or paracrine-signaling molecules. In many cases, they initiate their effects via interaction with extracellular GPCRs to affect downstream signaling pathways (Breyer & Breyer, 2000).
2.4 Prostaglandin and HSCs
PGE2 is the most abundant eicosanoid and has a variety of salutary effects in hematopoietic cells. In 1974, PGE2 was shown to regulate myeloid progenitor differentiation (Feher & Gidali, 1974). In the bone marrow, myeloid cells, as well as osteoclasts in particular, contain a high PGE2 synthetic capacity. Osteoclasts are an important component of the HSC niche and PGE2 released from these cells may regulate HSC and HPC function. PGE2 specifically binds to EP1–4 receptors, which are coupled to their respective G proteins to mediate distinct downstream pathways (Sugimoto & Narumiya, 2007). While EP3 receptor activation inhibits adenylate cyclase, EP2 and EP4 receptors activate it, which results in cAMP elevation. PGE2 dose-dependently inhibits growth of human and colony-forming units granulocyte/macrophage (CFU-GM) in vitro (Pelus, Broxmeyer, Kurland, & Moore, 1979; Pelus, Broxmeyer, & Moore, 1981) and myelopoiesis in vivo (Gentile, Byer, & Pelus, 1983) but stimulates erythroid and multilineage progenitor cells (Lu, Pelus, & Broxmeyer, 1984; Lu et al., 1987). Short-term ex vivo treatment of bone marrow cells with PGE2 increases the proportion of mouse colony-forming units spleen (CFU-S) (Feher & Gidali, 1974). In humans, PGE2 treatment increases the proportion of cycling CFU-GM from quiescent cells that appear to be stem cells. This effect is critically dependent on timing, duration of exposure, and concentration (Pelus, 1982).
Signal transduction after PGE2 engages in EP receptor can be either positive or negative, depending upon cell type. Positive signal transduction receptors include EP1, which triggers Ca2 + mobilization, and EP2 and EP4, which stimulate cAMP production. EP3 reduces cAMP concentration. PGE2 also enhances survival, proliferation, and homing of HSCs. Brief exposures to dmPGE2, a long-acting derivative of PGE2, ex vivo increase HSC frequency in murine bone marrow cells and enhance recovery of hematopoiesis in zebrafish following sublethal irradiation (Hoggatt, Singh, Sampath, & Pelus, 2009; North et al., 2007). Based on these promising preclinical studies, dmPGE2 is being investigated in phase II clinical trials for patients with leukemia receiving umbilical cord blood transplantation to expand and improve engraftment of HSCs (Cutler et al., 2013; Goessling et al., 2011).
Hematopoietic progenitors show a competitive advantage in repopulation assays after a brief dmPGE2 treatment, due to an increase in the homing, survival, and proliferation of HSCs (Hoggatt et al., 2009). The increased homing capacity of HSCs was due to enhanced expression of the chemokine receptor, CXCR4. CXCR4 is a receptor for the chemoattractant stromal cell-derived factor-1α (SDF-1α)/CXCL12, which has a key role in the trafficking and homing of HSCs and HPCs to the bone marrow niche. The CXCL12/CXCR4 axis plays an important role in progression of various tumors as CXCR4 increases metastasis of tumor cells into various organs, and CXCL12 can support the survival and proliferation of tumor cells. Notably, expression of CXCR4 is a poor prognostic marker in AML.
In addition, PGE2 signaling through the EP4 receptor increases β-catenin signaling, suggesting synergistic cross talk between prostaglandin and Wnt pathways (Wang, Mann, & DuBois, 2004). This is consistent with the finding showing PGE2-mediated increases in cAMP associated with β-catenin stabilization in HSC promoting survival. The in vivo significance of the PGE2/Wnt interaction is that HSC survival is promoted (Goessling et al., 2009). cAMP elevation by forskolin phenocopied the increase in HSC proliferation as well as demonstrated the requirement for Wnt/β-catenin. This PGE2 pathway might also be relevant in AML as downregulation of β-catenin by short hairpin RNA in both AML cell lines and AML blasts reduced proliferation of AML (Siapati et al., 2011).
2.5 MRP4 and prostaglandins
The understanding of how prostaglandins are extruded and taken up into cells is only a recent development because the conventional view was that these molecules entered and left cells by diffusion. Organic anion transporters mediate the uptake of prostaglandins (Schuster, 2002). The Borst lab was the first to demonstrate an ABC transporter (MRP4) actively export PGE1 and PGE2 (in the absence of additional cofactors, e.g., glutathione; Reid et al., 2003; Fig. 2). It is interesting that nonsteroidal anti-inflammatory drugs potently inhibit MRP4 and therefore the export of these pro-inflammatory prostaglandins. Cellular retention of PGE2 is related to MRP4 level with greater expression producing greater reductions in intracellular PGE2 concentrations (Reid et al., 2003). PGE2 export is blocked by inhibition or knockdown of MRP4 (Reid et al., 2003). Furthermore, MRP4 deficiency and RNA interference-mediated MRP4 knockdown significantly reduced extracellular PGE2. cAMP-dependent protein kinase activity and COX-2 expression are reduced in MRP4-deficient cells, suggesting that prostaglandin synthesis might be restrained along with a lack of prostaglandin transport (Lin et al., 2008). It is likely that MRP4-mediated export of PGE2 engages the EP receptors to modulate cAMP signaling and restrain prostaglandin synthesis. One could speculate that in the absence of MRP4, PGE2 levels in the bone marrow niche might be lower, producing reduced HSC function and/or homing. Furthermore, in HSC, MRP4 absence might also reduce PGE2 activation of EP2 and EP4, thereby reducing intracellular cAMP and producing a net reduction in HSC numbers.
2.6 Leukotrienes in hematopoietic cells
The leukotrienes are biosynthesized by oxygenation of AA by 5-lipoxygenase (5-LOX) and converted into the unstable intermediate LTA4 (Fig. 2). LTA4 is either enzymatically hydrolyzed to LTB4 or conjugated to glutathione forming the cysteinyl leukotriene LTC4. LTC4 is then converted to LTD4 and LTE4. Leukotrienes, like other eicosanoids, are mostly produced in myeloid cells (Lindgren & Edenius, 1993). 5-LOX is found in HSC (Bautz, Denzlinger, Kanz, & Mohle, 2001) and myeloid cells; however, loss of the Alox5 gene does not affect normal HSC function (Chen, Hu, Zhang, Peng, & Li, 2009). Notably, the leukotrienes LTB4, LTC4, and LTD4 increase the number of HSC both in mouse and in human (Braccioni et al., 2002; Elsas et al., 2008; Vore, Eling, Danilowicz, Tucker, & Luster, 1989). For LTB4, the increase in HSC is likely due to an increase in proliferation as well as a reduction in apoptosis (Chung et al., 2005).
In the BCR–ABL-mouse model of CML, 5-Lox/Alox5 is upregulated in LSCs. Interestingly, the increased 5-Lox activity is accompanied by an elevated plasma level of LTB4 (Chen et al., 2009). In a mouse model of CML, BCR–ABL failed to develop CML when bone marrow cells lacking Alox5 were used. The LSCs exhibited impaired differentiation, division, and displayed increased apoptosis (Chen et al., 2009). Accordingly, 5-LOX inhibitor, Zileuton, was more effective than the conventional therapeutic in prolonging the survival of CML mice. LTB4 has been implicated in reducing HSC self-renewal capacity and favoring differentiation (Chung et al., 2005); however, its precise role is not yet defined. Although it is not clear how Alox5 specifically regulates LSC, loss of Alox5 correlated with reduction of β-catenin expression in BCR–ABL-expressing hematopoietic cells. Furthermore, Alox5 function has been implicated in many signaling pathways including PI3K, which has an important role in AML.
2.7 MRP1 and leukotrienes
MRP1 was the second major drug transporter to be identified by the laboratories of Cole et al. (1992). MRP1 overexpression confers resistance to many drugs including anthracyclines and vinca alkaloids. MRP1 can act as a cotransporter with glutathione (GSH) of amphipathic organic anions, as well as an exporter of glutathione-, glucuronate-, or sulfate-conjugated drugs. Therefore, it is not surprising that the high-affinity endogenous substrate for MRP1 includes the GSH-conjugated leukotriene, LTC4. Using membrane vesicles prepared from MRP1 overexpressing HeLa cells, it was shown that MRP1 mediates ATP-dependent transport of LTC4, LTD4, and LTE4, with the highest affinity being for LTC4 (Leier et al., 1994).
A biological role for MRP1 as a LTC4 transporter in vivo was shown in the Mrp1-null mouse model (Wijnholds et al., 1997). Higher LTC4 concentrations were discovered in mast cells from Mrp1-null mice, but there was no obvious hematopoietic defect. Importantly, a dramatically attenuated LTC4-mediated inflammatory response was observed in mast cells from the Mrp1-null mice. These results showed that LTC4 export required MRP1 to mediate a normal mast cell inflammatory response. Accumulation of intracellular LTC4 resulted in increased LTB4 due to product inhibition of LTC4 synthase (Schultz, 2001). It is interesting to note that MRP4 has been shown to transport LTB4 in the presence of GSH (Rius, Hummel-Eisenbeiss, & Keppler, 2008). It is unknown if cross talk between MRP1 and MRP4 plays a role in inflammatory responses. However, because leukotriene biosynthesis is critical for LSC survival and self-renewal, LTC4 export by MRP1 might play a role in mediating signaling in leukemia cells (Fig. 2).
2.8 Porphyrin and ABCG2
One of the physiologically relevant substrates for ABCG2, pheophorbide a, a plant-derived chlorophyll metabolite, was identified using an Abcg2-null mouse model (Jonker et al., 2002). Although pheophorbide a is a chlorophyll metabolic breakdown product, it is a tetrapyrrole and structurally resembles endogenous porphyrins such as heme and protoporphyrin IX (PPIX). Heme is an essential cofactor to many proteins that regulate cell proliferation, death, and differentiation. Heme synthesis begins in the mitochondrial matrix with glycine and succinyl-CoA forming δ-aminolevulinic acid (ALA). Four enzymatic steps convert ALA to coproporphyrinogen III in the cytoplasm. The mitochondrial ABC transporter ABCB6 mediates the ATP-dependent import of coproporphyrinogen III back into the mitochondria where it is converted further to PPIX (Krishnamurthy et al., 2006; Lynch, Fukuda, Krishnamurthy, Du, & Schuetz, 2009). Iron is then enzymatically inserted into this tetrapyrrole to produce heme. Although heme is critical for multiple cell functions, its precursors, porphyrins, can be photoactivated to generate reactive oxygen species from molecular oxygen (see review by Ishikawa and colleagues, Chapter 7, Critical role of ABCG2 in ALA-photodynamic diagnosis and therapy of human brain tumor). In addition, PPIX can also induce cell death independent of photoactivation (Bednarz, Zawacka-Pankau, & Kowalska, 2007). The mechanism accounting for this nonphotoactivatable cell death is unknown but might be due to alterations in mitochondrial function. It is plausible that ABCG2 plays a role in modulating the excess intracellular PPIX to protect leukemic cells.
HSCs reside in a region of the bone marrow that has reduced oxygen level. The hypoxia-inducible factors (HIFs) are transcription factors, activated during hypoxia, that alter the metabolic pathways required for adaption to low oxygen environments (e.g., glycolytic enzymes). Hypoxia induces Abcg2 via HIFs (Krishnamurthy et al., 2004; Martin et al., 2008). The hematopoietic progenitors from Abcg2 KO mice exhibited reduced self-renewal of myeloid progenitors under hypoxia and chemical inhibition of ABCG2 in wild-type (WT) progenitors produced the same impairment in self-renewal. Conversely, overexpression of ABCG2 in a myeloid cell line (OCI-AML) resulted in increased hypoxic survival. The survival advantage conferred by ABCG2 was associated with its ability to export the heme precursor, PPIX (Krishnamurthy et al., 2004). During hypoxia, heme production is upregulated via increased ALAS and CPOX (Hofer, Wenger, Kramer, Ferreira, & Gassmann, 2003; Klinkenberg, Mennella, Luetkenhaus, & Zitomer, 2005; Vasconcelles et al., 2001). Therefore, ABCG2 may protect the hematopoietic and LSCs from cytotoxic PPIX over accumulation during hypoxia.
2.9 ABC transporters and AML
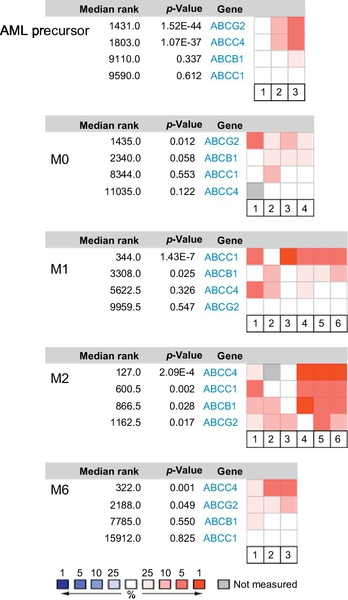
Much like normal hematopoiesis, AML blasts fall into a hierarchy, and depending on the progenitors stage, differentiation is arrested. AML can be divided into eight subtypes (M0–M7) according to the FAB classification based on the histopathology of the blasts (see Fig. 1). ABCG2 expression has been reported to correlate with an immature immunophenotype in normal hematopoietic cells, and its expression is downregulated upon differentiation of hematopoietic progenitors. One exception appears to be the erythroid-lineage which constitutively expresses ABCG2 (Raaijmakers, Van Den Bosch, Boezeman, De Witte, & Raymakers, 2002; Raaijmakers, van Emst, de Witte, Mensink, & Raymakers, 2002; Scharenberg, Harkey, & Torok-Storb, 2002; Zhou et al., 2002, 2001). We interrogated the Oncomine database (www.oncomine.org) to determine ABCG2 expression in various AML subtypes (Fig. 3). No significant expression was detected in myelocytic or monoblastic subtypes of AML (M3–M5). In contrast, ABCG2 was highly expressed in the myeloblastic leukemia subtypes (M0, without maturation) as well as in acute erythroleukemia (M6), a leukemia of red cell lineage. In pediatric AML M7, ABCG2 is highly expressed compared to other AML subtypes and a promoter unique to this subtype has been identified (Campbell et al., 2011).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


