Leukemia
The leukemias are a group of neoplastic disorders of the hematopoietic system, characterized by aberrant or arrested differentiation. The role of radiotherapy (RT) in conventional curative treatment is largely to deliver central nervous system (CNS) treatment in combination with systemic therapy for patients with acute lymphocytic leukemia (ALL). While the role of CNS prophylaxis has declined over the last 15 years because of toxicity concerns, it remains an important component of therapy for high-risk patients. Radiation oncologists must also be familiar with certain palliative situations unique to the leukemias as well as total body irradiation (discussed elsewhere), as the latter is often part of a stem cell transplant program.
Childhood ALL can now be cured in 80% to 89% of cases, with initial remissions generally occurring in about 90%.1–3 In adults, initial remission rates are generally equally high, but cure rates are only in the 30% to 45% range.4 In contrast to the situation in pediatric ALL, pediatric acute myeloid leukemia (AML) generally fairs less well, with 60% cure rates, although this represents a marked improvement since the 1970s when cure rates were roughly 20%.2 Adult AML cures are less frequent in the overall population, in large part a reflection that this disease principally afflicts older individuals. In the United States, the estimated annual incidence and mortality from AML are 12,330 cases and 8,950 deaths.2 Allogeneic stem cell transplantation has specifically targeted refractory/recurrent ALL and AML. Reduced-intensity transplants are being investigated to address the needs of high-risk or elderly patients with acute leukemia.
Chronic myelogenous leukemia (CML) and chronic lymphocytic leukemia (CLL) are primarily diseases of adults and have natural histories measured in years to decades. Both have the ability to transform to more aggressive diseases, though CML, in particular, reliably progresses to a more acute disease or blast crisis in its terminal phase. Allogeneic stem cell transplantation is associated with a high cure rate for patients with CML if conducted early in the chronic phase of the disease. However, due to the widespread use of the ABL tyrosine kinase inhibitors, the use of allogenic transplant for CML has declined dramatically over the past decade.5
FIGURE 80.1. Cranial irradiation field outlining treatment that encompasses the entire cranial subarachnoid space. The radio-opaque markers outline the anterior aspect of the bony orbit so as to demarcate inclusion of the posterior aspect of the eye within the treatment fields.
 ANATOMIC CONSIDERATIONS
ANATOMIC CONSIDERATIONS
The radiation oncologist must be knowledgeable about the anatomy of the CNS, particularly the meninges (dura mater, arachnoid, and pia mater) and subarachnoid space for the proper design of CNS-directed radiation treatments. The epidural space lies between periosteum and the dura, whereas the potential space between the dura and arachnoid is called the subdural space. The pia and arachnoid are often described as a combined membrane called the leptomeninges. Between the arachnoid and pia is the subarachnoid space, which is filled with cerebrospinal fluid (CSF). The pia hugs the surface of the brain extending into sulci, fissures, and the internal cavities or ventricles while the bulk of the arachnoid follows along the dura except for the fine trabeculations and into some of the major fissures of the brain. Arachnoid sheathes both nerves and blood vessels penetrating the pia and exiting the CNS, creating relatively short segments of subarachnoid space, which is of particular anatomical importance in the vertebral column. In the design of the inferior aspect of craniospinal radiation fields, the subarachnoid space extends laterally to the spinal ganglia, which are located within the intervertebral foramina.6,7 Thus, coverage of the entire sacroiliac joints in such spinal field design is excessive. The caudad extent of the subarachnoid space is variable within the sacrum.8 Clinically, the end of the thecal sac is now routinely determined on magnetic resonance image scanning. Pathologically, CNS leukemia originates as a perivascular infiltrate along the subpial blood vessels. As this progresses, the leukemia infiltrates preferentially into the subarachnoid space as well as into the brain parenchyma.9
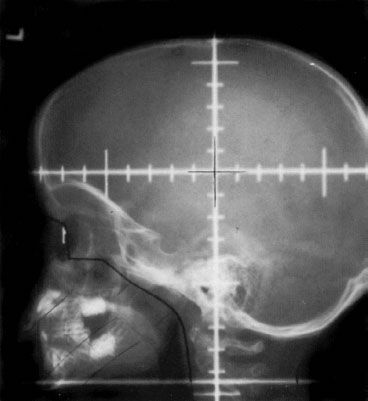
Base of skull anatomy is also of importance to the radiation oncologist in the design of lateral cranial fields. The middle cranial fossa and temporal lobe project over the sphenoid sinuses on lateral radiographs. The cribriform plate projects along the roof of the orbit along an imaginary line that connects to the inferior aspect of the frontal sinuses. Several radiologic-anatomic studies have pointed out how the eye’s lens in this “beam’s-eye view” is <1 cm away from the cribriform plate, having implications in field design. Insuring coverage of the entire subarachnoid space should supersede the concerns of radiation-induced cataracts.10,11 Moreover, because subarachnoid space extends as a sheath along the optic nerve, it is standard to include the posterior half of the eye globes within cranial radiation fields by using the anterolateral aspect of the bony orbit as an anatomic landmark, as it typically lies along a line that roughly bisects the eye11 (Figs. 80.1 and 80.2).
To compensate for the blood–brain barrier, certain drugs are administered directly into the subarachnoid space either via lumbar puncture or via an intraventricular reservoir (e.g., an Ommaya reservoir). Intrathecal (IT) drugs distribute unevenly, however, throughout the subarachnoid space.12,13 This is not surprising given the knowledge of brain anatomy and CSF circulation. CSF is mainly formed in the brain ventricles by the choroid plexuses, which are specialized capillary-rich tufted structures. This fluid then flows through the intraventricular foramina and the cerebral aqueduct of the midbrain into the fourth ventricle, from where it communicates with the rest of the subarachnoid space. CSF is resorbed principally in the dural sinuses via buttonlike projections called arachnoid villi. Thus, IT therapy theoretically undertreats the ventricular spaces and cerebral/cerebellar sulci as well as any gross disease extending into the brain substance. This concern has led to the concept of combining cranial RT with IT chemotherapy; the latter to cover the spinal subarachnoid space. Similarly, when craniospinal irradiation (CSI) is needed to treat a higher burden of CNS leukemia, IT therapy allows the spine to be treated to a lower dose than the brain.
FIGURE 80.2. Skull views outlining anatomic limits of the base of skull for inclusion of the cranial subarachnoid space. A: Radio-opaque markers have been placed at the cribriform plate (C), roof of the orbit (O), and the temporal fossa (T). B: Plain lateral radiograph shows the projection of the cribriform plate, orbital roof, and temporal fossa.
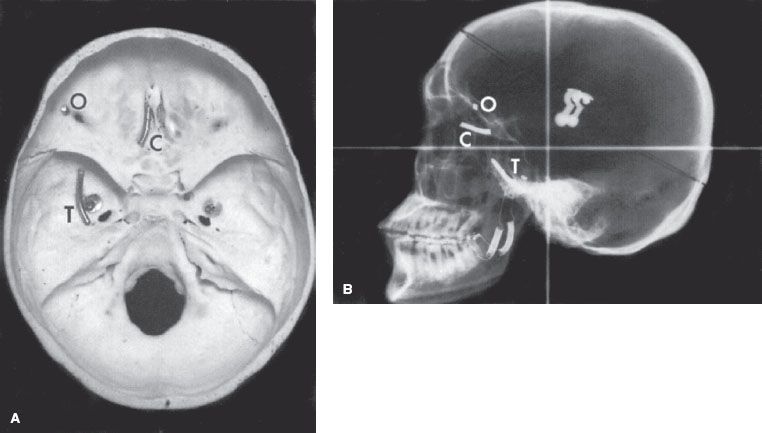
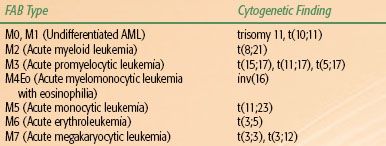
TABLE 80.1 CORRELATION OF FRENCH-AMERICAN-BRITISH (FAB) CLASSIFICATION OF ACUTE MYELOGENOUS LEUKEMIA (AML) WITH COMMON CYTOGENETIC ABNORMALITIES
 ACUTE LEUKEMIAS
ACUTE LEUKEMIAS
Classification Systems, Pathology, and Risk Stratification
Acute leukemias have traditionally been classified using the French-American-British (FAB) morphologic criteria. AML and its subtypes can most often be identified microscopically by the presence of Auer rods, staining for myeloperoxidase or monocyte-associated esterases, and other cytologic features of differentiation. Certain chromosome translocations are common to each AML subtype (Table 80.1). AML with minimal or no myeloid differentiation (M1 and M0 subtypes) can be confused with ALL but for flow cytometric identification of early myeloid antigens. The more differentiated AML subtypes M2-M6 are usually recognizable by morphology and cytochemistry.
The FAB assignment of subtypes is increasingly being replaced by the World Health Organization (WHO) classification scheme for myeloid neoplasms.14 The WHO classification creates four key subgroups: AML with recurrent genetic abnormalities (including t(8;21), inv(16), t(15;17), t(9;11) among others), AML with myelodysplasia-related changes, therapy-related neoplasms, and AML not otherwise specified. The new subgroups are meant to highlight meaningful biologic and genetic differences between disease entities with differing prognoses and clinical behavior. In addition, in the WHO scheme, the number of blasts in the blood or bone marrow required to confirm a diagnosis of AML is 20%, instead of the 30% specified by the older FAB criteria.
The FAB classification for ALL specified three subtypes based on morphology, which is largely of historical interest. L1 is the predominant type in 85% of childhood ALL. It is characterized by small cells with scanty cytoplasm and inconspicuous nucleoli. L2 is common in ALL of adults and is identified morphologically by blasts that show prominent nucleoli, abundant cytoplasm, and more variability in size. L3 lymphoblasts are large cells with cytoplasmic basophilia and vacuolization, similar to Burkitt’s lymphoma cells. The common ALL antigen (CALLA) is expressed in about 85% of ALL cases. At least with childhood ALL, this FAB system has not proved to be terribly useful with high interobserver variability and a lack of correlation with the more prognostically important immunologic and genetic features for ALL.15 In the WHO classification, most of these entities, with the exception of Burkitt’s, fall under the category of precursor lymphoid neoplasms. These are subcategorized into B-cell lymphoblastic leukemia/lymphoma, not otherwise specified; B-cell leukemia/lymphoma with recurrent genetic abnormalities; and T-lymphoblastic leukemia/lymphoma.16
As may be apparent from the above discussion of the transition from the FAB to the WHO criteria for categorization, immunophenotyping and molecular markers have become very important in the classification of leukemia. Flow cytometry using specific monoclonal antibodies can be used to assay for a panel of antigens, often known as cluster of differentiation (CD) molecules, that define leukocyte maturation. For instance, CD19 and cytoplasmic CD79a define B-cell lineage, whereas CD7 and CD3 define T-cell lineage. Myeloid cells are correlated with positivity for CD13, CD33, and cytoplasmic myeloperoxidase. With other markers defining different degrees of differentiation and maturation, ALL is now conventionally divided into T-cell and B-cell lineage. B-cell leukemia has four distinct subclasses: early pre-B, pre-B, transitional pre-B, and the more mature B-cell. The transitional pre-B subclass is relatively new and is associated with a relatively good prognosis in children.17
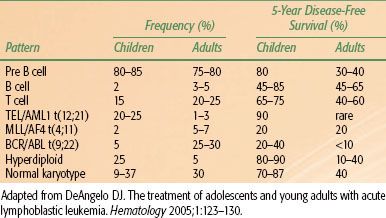
Cytogenetic or chromosomal abnormalities occur in up to 90% of ALL cases. Of these, roughly two-thirds are nonrandom, falling into distinct patterns. Some of these have distinct prognostic and therapeutic implications. Adult and childhood forms of ALL have distinctly different patterns of genetic abnormalities as well as immunophenotyping (Table 80.2). This may partially explain the poorer prognosis in older patients. For example, the Philadelphia chromosome or bcr-abl gene fusion/t(9;22) translocation in precursor B-cell ALL is associated with a poorer prognosis. It occurs in 4% of childhood ALL cases compared to about 25% in adults, but it now represents an important therapeutic target with the development of tyrosine kinase inhibitors. Abnormal DNA ploidy is extremely common, but two patterns seem to be clinically important. Hyperdiploidy with >51 chromosomes per cell (or DNA index >1.6) occurs in 25% of children with ALL. Although there is some association with favorable clinical factors (age >10 years and low presenting leukocyte counts), hyperdiploidy is an independent favorable factor; however, in adults with ALL, outcomes are poor even in presence of hyperdiploidy. In childhood ALL, trisomies 4, 10, and 17 are associated with a good prognosis. Similarly, TEL/AML1 fusion gene expression (cryptic t(12;21) translocation, is a common translocation, occurring in 15% of pediatric ALL patients, in which an in utero event juxtaposes the RUNX1 gene on chromosome 21 with the ETV6 gene on chromosome 12. The resulting chimeric protein impairs normal hematopoietic differentiation, increasing the self-renewal capacity of early progenitor cells. Distinct poor molecular prognostic factors in pediatric ALL include not only the aforementioned Philadelphia chromosome but also MLL-AF4 or t(4;11), which primarily occurs in infants as well as hypodiploidy (<44 paired chromosomes).
The aforementioned laboratory assessment and clinical disease features have led to distinct risk stratification categories, at least as far as childhood ALL is concerned. Clinical prognostic features of B-cell leukemias have been commonly used to place patients into risk groups. Using the National Cancer Institute’s (NCI) risk classification scheme, standard risk includes patients (two thirds of pediatric B-cell ALL patients) whose age at diagnosis is between 1 and 10 years as well as a presenting leukocyte count of <50,000/μL, in the absence of CNS involvement, was historically associated with an 80% 4-year disease-free survival.18 High-risk patients—defined as a high white blood cell (WBC) count, age below 1 year or above 10 years, or CNS involvement at diagnosis—had a corresponding 65% 4-year disease-free survival. T-cell phenotype occurs in 12% to 15% of pediatric ALL cases and was once thought to convey a relatively poorer prognosis. This prejudice has been due to numerous unfavorable clinical correlations, including older age, male sex, elevated WBC count, extensive extramedullary disease including mediastinal and peripheral adenopathy or hepatosplenomegaly, and a higher tendency for relapse in the CNS and testes in males.19 Current clinical practice is for T-cell leukemias to be treated on different protocols than B-lineage ALL. Nevertheless, when one factors out unfavorable clinical features, the prognostic difference between T- and B-cell ALL is difficult to discern. In regards to B-cell ALL, current protocols assign patients to low, standard, and high risk. Low-risk patients are operationally defined by standard risk features along with a rapid early response to induction chemotherapy.20 Age is also an important prognostic factor. Apart from this, the intensity of the treatment program is of importance as shown by the fact that adolescents treated on pediatric leukemia regimens have had better outcomes than those treated on adult programs.21
TABLE 80.2 FREQUENCY AND DISEASE CONTROL ASSOCIATED WITH IMMUNOPHENOTYPES AND CYTOGENETIC ABNORMALITIES AND SURVIVAL IN ACUTE LYMPHOBLASTIC LEUKEMIAIN CHILDREN (AGES 1–18) VERSUS ADULTS (AGES 18 AND OLDER)
The response to induction chemotherapy has also been proven to be an extremely important prognostic category. In pediatric ALL, the concept of minimal residual disease (MRD) detected by flow cytometry of postinduction peripheral blood or marrow aspirates has led to critical prognostic stratification. A contemporary analysis of MRD in a pediatric cooperative group trial showed that even up to 0.01% blasts from day 8 postinduction mononuclear peripheral blood cells conferred a poorer prognosis.22 Moreover, day 29 and end of consolidation bone marrow MRD was able to further segregate unfavorable patient subgroups taking into account other prognostic factors such as TEL/AML1, trisomies 4 and 10, and NCI risk class. From the assessment of MRD, the concept of risk-adapted therapy has allowed for improved outcomes with the intensification of therapy in those patients who continue to have detectable lymphoblasts after initial chemotherapy.
Assessment of CNS involvement at diagnosis in acute leukemia is critical as to risk stratification, which is of course of particular importance to the radiation oncologist. Unfortunately, a traumatic lumbar puncture has been noted to be associated with a worse prognosis.23 Regardless, CNS involvement has been unequivocally defined by a CSF leukocyte count of ≥5 WBC/μL along with either blast cells on cytospin or the presence of cranial nerve palsy. By convention, this is now classified as CNS-3. CNS-1 is defined as no blast cells on CSF cytology, whereas CNS-2 is <5 WBC/μL with blast cells present. Clinical data is conflicting as to the prognostic importance of CNS-2, reflecting the fact that CNS involvement is not simply a distinction of being present or absent, but that it has also been associated with improved control with more intensive systemic and IT therapies.24,25 Patients with CNS-3 disease who have more intensive IT chemotherapy along with cranial radiation within the first year of therapy have similar event-free survival as CNS-2 patients. For patients in remission who undergo surveillance lumbar punctures, there is even controversy regarding the prognostic importance of finding low numbers of blast or atypical cells in the CSF.26,27
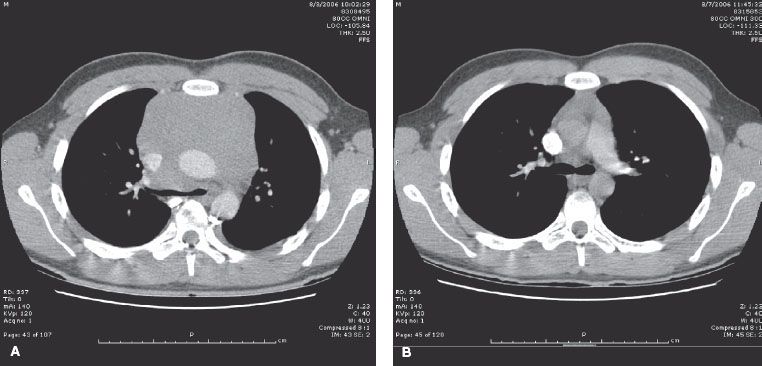
FIGURE 80.3. Computed tomography scans of chest before (A) and after (B) palliative radiotherapy (RT) for mediastinal mass associated with T-cell acute lymphocytic leukemia. A 16-year-old boy presented with dyspnea and chest pain. A chest x-ray showed a mediastinal mass while blood counts revealed a lymphocytosis. Diagnosis was established by flow cytometry of blood. Initial treatment with steroids, doxorubicin, vincristine, and methotrexate failed to produce immediate response. Megavoltage RT with anterior and posterior opposed beams for 6 Gy in three fractions was administered, subsequently relieving acute symptoms and allowing for general anesthesia to take place without risk for complete airway obstruction in order for central line placement and lumbar puncture to be performed.
Radiotherapeutic Emergencies
The radiation oncologist will be called on to assist with certain emergencies when the patient first presents with leukemia or at the time of relapse. Mediastinal adenopathy causing airway compression or spinal cord compression from epidural disease are clear indications for emergent RT. Generally, only one to three 1.5- to 2.0-Gy fractions are required while the diagnosis is being established and systemic therapy is being initiated (Fig. 80.3). Glucocorticoids are an important adjunct to CNS RT but can produce rapid lysis of some lymphoblastic lymphomas/leukemias, which may hamper diagnostic evaluation. In the presence of cranial nerve palsies at diagnosis, some radiation oncologists recommend 10 to 15 Gy to the base of skull early in the treatment course to try and reverse the neurologic deficits.28,29 Extreme leukocytosis with blast counts over 75,000 to 100,000/μL is a concern with myeloid leukemia, as leukostasis may occur, particularly in the vessels of the brain or lung. Lymphoid blasts are less adhesive to vessel walls and blast counts of up to 400,000/μL or more are often well tolerated. In decades past, RT directed at the whole brain was employed using low doses on the order of 6 to 10 Gy in various fractionations. However, the role of RT in this setting has been questioned,15,30,31 but may be considered when leukophoresis or exchange transfusion is contraindicated or unavailable.
Treatment of Acute Myeloid Leukemias
Classical induction therapy for AML is an anthracycline on days 1 to 3 with cytarabine (cytosine arabinoside, ara-C) for 7 days. Acute promyelocytic leukemia (M3 AML, APL) represents an exception to this rule. In many cases, the disseminated intravascular coagulation and associated PML-RAR fusion in APL are best managed with a combination of anthracycline and all-trans-retinoic acid or single-agent arsenic trioxide.32 For the remainder of the AML subtypes, daunorubicin and idarubicin are the anthracyclines most commonly used in induction chemotherapy regimens. Although a meta-analysis of multiple trials suggested that idarubicin has a higher complete response (CR) rate and survival over daunomycin,33 more recent data from dose escalation trials using daunorubicin as part of the induction regimen suggest that when equivalently dosed, responses are similar.34 Remission rates vary from 50% to 80% dependant on patient age, karyotype, and subtype of AML. Patients younger than age 60 have CR rates of 70% to 80%, whereas older patients tend to have lower CR rates of 50% to 60%. Patients who develop secondary AML following chemotherapy for other cancers have CR rates in the 40% to 60% range. Some induction regimens in children have added other drugs such as etoposide. Adding other chemotherapy drugs to anthracycline and cytarabine has yet to be shown to produce a convincing survival benefit for AML induction in adults.35 Adults older than 60 years of age with AML and intermediate or unfavorable cytogenetics who are not candidates for hematopoietic stem cell transplantation are generally considered suitable for treatment with low-intensity palliative therapies such as low-dose subcutaneous cytarabine, azacitidine, or decitabine.36 Alternatively, such patients may be referred for clinical trials of novel agents.
Once remission is achieved, the need for additional therapy has been well documented in large randomized trials. Current data suggests that optimal postremission consolidative therapy begins with high-dose ara-C for up to four cycles.37 Alternatively, additional cycles of anthracycline for 2 days along with conventional-dose cytarabine for 5 days has been used in both younger and older individuals with AML. Lumping all types of AML together as one group, the roughly 10% to 20% of patients who then remain in remission for 3 years have a high likelihood of being cured. High-dose ara-C has considerable CNS toxicity in elderly patients and has also been avoided in pediatric protocols. Other drugs used for consolidative treatment or for second-line induction therapy include etoposide, 6-mercaptopurine, amsacrine, 5-azacytidine, and methotrexate (MTX), but no one regimen is clearly advantageous.
The role of CNS prophylaxis is not well defined for AML, particularly because CNS relapse rates are relatively infrequent (at roughly 5% to 10%). Some studies show no difference in relapse rates with cranial radiation. Nevertheless, many pediatric regimens employ IT drugs such as ara-C. Patients with a high WBC count at diagnosis or monocytic variants of AML are believed to have a higher risk for CNS relapse, which may justify both IT chemotherapy and cranial radiation in this setting. At least one study of childhood AML demonstrated a lower systemic relapse rate after cranial radiation.38
Leukemia-free survival rates vary widely from 15% to 80% in AML and are dependent on risk stratification. Younger patients with standard risk M2 AML or with favorable cytogenetic abnormalities have relatively better prognoses, whereas those with older age, induction failure, adverse cytogenetic abnormalities, or other FAB subtypes of AML do worse.39,40 An exception is acute promyelocytic leukemia, which is among the most curable types of AML thanks to the efficacy of all-trans-retinoic acid (ATRA) in the management of this leukemic subtype. Postremission therapy for AML patients with adverse features may include transplantation with autologous or allogeneic stem cells.
The radiation oncologist may be called on to treat myeloid or granulocytic sarcoma, which may predate a diagnosis of leukemia or be a harbinger of systemic relapse, and are typically associated with AML, but may also be seen with myeloproliferative disorders.41,42 Also called a chloroma, these solid masses of leukemic infiltrates are responsive to modest doses of RT. The name chloroma derives from the fact that myeloid cells contain myeloperoxidase, which manifests a greenish color on gross inspection. These may occur in all varieties of extramedullary sites including periosteum, skin, soft tissues, gastrointestinal tract, the spine, and in epidural spaces or meninges.43–46 Pathologic misclassifications are common, as this is a rare clinicopathologic disorder. Based on various case reports in the literature, symptomatic problems from chloromas may be readily relieved with doses of 10 to 20 Gy. The fractionation and total dose needs to take into consideration any normal tissue toxicities and the potential for future total body irradiation (TBI) should the patient be a potential candidate for an allotransplant. Although overall survival may be better than AML in many chloromas, long-term disease control usually requires systemic therapy used for AML.41,47–48,49,50
Treatment of Acute Lymphoblastic Leukemia
The four components of specific ALL therapy are (i) induction of remission, (ii) intensification and/or consolidation, (iii) maintenance therapy, and (iv) CNS prophylaxis. In the 1960s, the problem of CNS recurrence was addressed with CNS radiation and IT chemotherapy. Subsequent improvements in ALL cure rates over the last 2 to 3 decades has resulted from improved risk stratification of patients with more effective, tailored multidrug regimens. As discussed in the following text, the late sequela of 24-Gy cranial irradiation were recognized in the 1980s and 1990s, leading to the elimination of cranial RT in favor of intermediate- or high-dose MTX in all but high-risk patients or those with CNS-3 disease. While not a standard worldwide, investigators at St. Jude Children’s Research Hospital (SJCRH) have completely eliminated the use of upfront cranial RT in their protocols of pediatric ALL, accepting a small incidence of CNS relapse in their high-risk patients.51 In addition, with the improvements in systemic therapy, testicular relapse in males has become a rare event.52 Testicular irradiation is rarely necessary except in the setting of testicular relapse or bone marrow transplantation.
Induction therapy for ALL as a minimum typically includes a glucocorticoid, vincristine, and L-asparaginase. Higher-risk patients often receive additional drugs such as an anthracycline, especially for adult ALL. Other four or greater multidrug regimens have been used, all of which has resulted in initial remission rates of 95% to 99% in children and 75% to 90% in adults. Dexamethasone is now the preferred systemic steroid treatment as suggested in two clinical reports comparing it to prednisone, with dexamethasone reducing both CNS and systemic relapse in pediatric ALL.53,54 This may be related to dexamethasone’s better penetration into the CSF and longer half-life, providing enhanced protection against CNS relapse.55
Following induction and achieving CR, patients then receive intensification therapies that have been developed by cooperative groups in the United States and Europe.56,57 This may consist of high doses of antimetabolites such as MTX, ara-C, or L-asparaginase. Additional anthracycline therapy is beneficial in high-risk patients.58 It is believed that high-dose MTX helps control CNS disease, which has allowed for less use of cranial radiation in some pediatric programs.
Following intensification therapy, patients undergo a phase of maintenance treatment. In all but mature B-cell ALL, maintenance therapy over 2 to 3 years with agents such as weekly low-dose MTX and mercaptopurine appears to be quite important, albeit for unclear reasons. In high-risk patients, cooperative group studies have demonstrated a benefit to an additional “delayed” intensification after a period of maintenance therapy.56,59
For ALL in adults, particularly those with the L3 subtype, patients have experienced improved remission rates with multiagent induction regimens that include high-dose MTX, cyclophosphamide, and sometimes ara-C. These drugs in combination with vincristine, steroids, L-asparaginase, and sometimes doxorubicin have been associated with 68% to 85% CR rates.
For relapsed ALL, a second induction generally consists of multiple drugs including vincristine, prednisone, L-asparaginase, an anthracycline with or without MTX, VP-16 or teniposide, and ara-C. If the CNS is involved, IT treatment is given along with RT.60 If testicular relapse occurs, bilateral testicular irradiation is administered. When isolated CNS or testicular relapse develops, systemic therapy is indicated along with local RT.61
If a second remission develops, as occurs in 70% to 90% of cases, subsequent treatment includes either allogeneic transplantation or consolidative chemotherapy.61,62 There are no randomized trials to indicate which is better, but comparative analysis suggests that survival is improved with allogeneic transplantation, particularly in ALL patients who had brief initial remissions.62,63
CNS Prophylaxis of Acute Leukemia and the Role of Cranial Radiotherapy
Historically, as multiagent chemotherapy proved to be highly effective in producing remissions in childhood ALL, numerous investigators noted a significant increase in CNS relapses. The CNS was recognized as a sanctuary site, protected from chemotherapy by the blood–brain barrier. In addition, CNS recurrences invariably led to systemic recurrence suggesting that CNS disease was capable of reseeding the blood and marrow.
This observation led to a long series of CNS preventative therapy trials, which initially utilized craniospinal irradiation (CSI). For instance, studies V and VI in 1962–1967 from SJCRH established that CSI to 24 Gy in 15 to 16 fractions reduced the isolated CNS relapse rate from 67% to 4%.64,65 CSI doses of 12 Gy did not appear to be effective with the early chemotherapy regimens from this time.66 Concerns that full spinal RT would be associated with more acute myelosuppression, late musculoskeletal hypoplasia, as well as the technical difficulties of CSI, led SJCRH investigators to compare CSI with cranial radiation plus IT MTX in study VII.67 Here, the two CNS preventative regimens were found to be equivalent with roughly an 8% risk of CNS relapse.
In SJCRH study VIII (1972–1975), all patients received 24-Gy cranial radiation plus IT MTX. Patients were randomized to one of four maintenance regimens: (i) weekly intravenous (IV) MTX begun during cranial radiation, (ii) oral MTX and 6-mercaptopurine, (iii) oral MTX, mercaptopurine, and cyclophosphamide, and (iv) same three drugs plus ara-C.68 The incidence of CNS relapse was 5.0%, 1.5%, 20%, and 11.4%, respectively. More troublesome, however, was the development in some patients of leukoencephalopathy, a disabling syndrome of lethargy, seizures, spasticity, paresis, and ataxia. The incidence of leukoencephalopathy in the four randomization groups was 55%, 0%, 7.1%, and 1.4%, respectively. Thus, standard maintenance with oral MTX and mercaptopurine following CNS treatment with cranial radiation along with IT MTX to treat the spinal subarachnoid space was found to have the lowest CNS relapse rate and the least toxicity. The major lesson learned was that IV MTX and cranial radiation in close temporal proximity should be avoided.
In the 1970s and 1980s, additional phase III trials further defined appropriate preventative CNS therapy for childhood ALL. The Children’s Cancer Study Group (CCSG) trial #101 compared: (i) 24-Gy CSI plus extended field RT encompassing the liver, spleen, and gonads; (ii) 24-Gy CSI alone; (iii) 24-Gy cranial RT plus IT MTX; and (iv) IT MTX alone. Overall, the different radiation regimens were comparable in preventing CNS relapse while statistically superior to IT chemotherapy alone.69 This finding was further confirmed in a cross-study comparison70 as well as a Cancer and Leukemia Group B trial.52 The CCSG further compared cranial RT plus IT MTX with CSI in high-risk patients, defined by a WBC at diagnosis of >50,000/μL; cranial irradiation and IT MTX proved to be significantly superior with respect to both CNS and systemic relapse rates.
Further cooperative group trials have refined the efficacy of CNS-directed therapies within patient groups stratified by risk. Several trials have compared cranial RT with intermediate- or high-dose IV MTX along with IT chemotherapy (some with the addition of IT hydrocortisone and ara-C to MTX know as “triple” IT therapy).52,71,72 The essential findings have been that in patients with low- or standard-risk ALL (e.g., age 3 to 6 years and WBC count <10,000/μL) who are managed without cranial radiation by substituting IT MTX throughout induction, consolidation, and maintenance therapy, CNS relapse rates have remained at a low level of 5% or less.73–75 Additionally, with triple IT chemotherapy, there was a trend toward fewer systemic failures as well as excellent CNS control when given throughout consolidation and maintenance therapy. This may be at the expense of increased neurotoxicity, however, when given in conjunction with intermediate-dose IV MTX.75
Moreover, a meta-analysis of 65 randomized trials of pediatric ALL worldwide initiated prior to 1993 that evaluated the role cranial RT concluded that in the vast majority of patients, cranial RT may be avoided with the use of extended IT therapy.76 IV MTX was particularly advantageous in reducing systemic relapse. One notable exception to the conclusions of aggregated trials of this meta-analysis was the CCG-105 trial of intermediate-risk ALL. In CCG-105, 1,388 patients (with a complex matrix of adverse risk factors defined by patient age, presenting WBC count, and FAB subtype) were randomly assigned to receive either IT MTX alone or cranial radiation for CNS treatment.77 A secondary complex randomization scheme allocated patients to standard or intensive chemotherapy. Intensive chemotherapy included either more drugs for induction or the addition of a delayed intensification chemotherapy phase after consolidative and CNS therapies. CNS recurrence rates were comparable in all groups at roughly 5% to 7% except in those patients receiving standard chemotherapy without cranial radiation, where the CNS recurrence was 20%. Thus, more intensive systemic therapy can lower CNS recurrence rates, abrogating any benefit to cranial irradiation. Finally, one interesting subanalysis of this systematic review of cranial RT for ALL looked at 809 patients across seven trials78,79–82 that compared different doses of radiation (generally 18 to 21 Gy versus 24 Gy). There were no differences in CNS or non-CNS relapse rates that could be discerned.
Regarding present-day use of prophylactic cranial RT, investigators have been focusing on patients who are at the highest risk for CNS relapse. At one extreme is the St. Jude’s group, which has become particularly concerned about the late effects of 24 Gy to the brain in terms of neurocognitive disabilities, hypothalamic/pituitary dysfunction, and secondary malignancies. With risk-adapted intensification of chemotherapy, Pui et al.51 have reported favorable results with the complete elimination of cranial RT in all newly diagnosed ALL patients.51 In their St. Jude Total XV trial, 498 patients were treated without cranial RT yielding an 86.6% 5-year event-free survival, which was comparable to their prior studies that had included cranial RT. CNS-directed therapy used five cycles of high-dose MTX including, as part of induction therapy, dexamethasone, and extended IT chemotherapy. High-risk ALL patients received up to 16 to 25 IT chemotherapy sessions, whereas low-risk patients received 13 to 18 doses of IT chemotherapy. Patients who were at increased risk of CNS relapse were those who had positive CSF cytology (CNS-2 or -3) or T-cell ALL. Moreover, a relatively high 7% of patients in first remission went on to undergo an allogeneic stem cell transplant correlating with high-risk features of MRD and adverse cytogenetic markers.
Elsewhere worldwide, the philosophy on the use of cranial RT in pediatric ALL has been to use it judiciously in high-risk patients while at the same time also reducing radiation doses to address the concerns about late effects, which includes learning disabilities and other cognitive defects, growth retardation, hypopituitarism, secondary malignancies, and the aforementioned leukoencephalopathy. The European BFM-ALL trials since 1990 have reduced the cranial radiation dose to 12 Gy with their risk-adapted intensification schemes, although those with CNS-3 disease received 24 Gy in their BFM-90 trial and 18 Gy in the more recent BFM-95 trial. Currently, the Children’s Oncology Group (COG) has followed suit with a reduction in cranial radiation doses in its current ALL trials.
High-risk B-cell ALL patients who continue to receive cranial radiation are generally defined by age and high presenting WBC counts per NCI criteria, by adverse cytogenetics (e.g., bcr-abl, MLL, and hypodiploidy), and by MRD after induction chemotherapy. Most T-cell ALL patients are at increased risk for CNS relapse due to accompanying risk factors and, therefore, continue to receive cranial irradiation. An analysis of T-cell ALL patients (generally with other poor-risk features) treated within several Pediatric Oncology Group (POG) protocols suggested that omitting cranial radiation had an adverse impact on CNS relapse rates.83 Specifically, the 3-year CNS relapse rate was 18% for those who did not receive RT compared to 7% who did. On the other hand, a European report looking at a subgroup of favorable T-cell ALL patients, generally those with young age and low WBCs, suggests that it may be safe to omit cranial radiation in this subgroup.73 However, as most T-cell ALL patients present with high WBC counts, Conter et al.73 published a relevant comparison of the AIEOP-91 trial with the BFM-90 trial in which similar backbone chemotherapy is used. The AIEOP-91 trial omits cranial RT albeit with more IT chemotherapy, leading to a significantly higher CNS relapse along with a 3-year event-free survival of 62% compared to 88% for the BFM-90 patients who received cranial RT. Multivariate analysis showed that age younger than 10 years and WBC >100,000 in particular defined a group of T-cell ALL patients who should receive cranial RT.
To reduce the toxicity of prophylactic cranial radiation, several investigations have explored a reduction in radiation dose. The use of 18 Gy in 9 or 10 fractions along with IT MTX yields comparable disease control rates as 24 Gy.84 Although there were some initial reports of reduced cognitive dysfunction even with such a dose reduction,85 this issue is by no means completely settled.86–88 Nevertheless, a dose of 18 Gy for prophylactic cranial irradiation had become more or less standard for pediatric ALL being treated off-protocol. As noted in the following text, however, patients being treated with BFM-type chemotherapy programs may be treated with lower cranial RT doses of 12 Gy. For adults with ALL, various protocols have used 24 Gy, whereas others employ 18 Gy.87,89 Some programs such as HyperCVAD omit the use of cranial RT in the face of high-dose MTX and ara-C, which effectively penetrate the blood–brain barrier. The Dana Farber Cancer Institute ALL Consortium has studied the role of hyperfractionated cranial radiation (0.9 Gy twice daily) compared to standard daily fractionated treatments (1.8 Gy daily) to 18 Gy in high-risk patients.90,91 Results from Dana Farber ALL 87–04 show excellent CNS control rates with both the standard or hyperfractionated treatments. Late neurocognitive sequela has been similar in which systemic therapy emphasizes L-asparaginase and omits high-dose MTX. A follow-up Dana Farber ALL trial 95-01, compared hyperfractionated 18 Gy cranial RT to extended IT chemotherapy without cranial RT. The interesting finding here is that quantitative measurements of neurocognitive function were similar in both groups.92
The German-Austrian-Swiss ALL-BFM (“Berlin-Frankfurt-Munich”) Study Group has further reduced the radiation dose to 12 Gy, initially in a selected group of standard-risk pediatric patients.74 With modern chemotherapy regimens as opposed to the older SJCRH experience, this further reduction in radiation dose was associated with excellent CNS control. Moreover, the ALL-BFM 90 protocol stopped using cranial RT in low- or standard-risk patients, but both the medium- and high-risk patients received 12-Gy prophylactic cranial radiation resulting in CNS recurrence rates well below 5%.58 For the ALL-BFM 95 trial, cranial RT was deleted for the intermediate-risk B-cell ALL patients. This has resulted in a small increased risk for CNS relapse, which is considered clinically acceptable in order to avoid the late effects of such RT.93 Specifically comparing intermediate-risk BFM 90 to BFM 95 patients, the isolated CNS relapse rate at 6 years is 0.5% versus 1.9%, p <0.01. The 6-year “any-CNS” relapse rate was also statistically different at 2.2% versus 4.4%. Further follow-up and additional experience within the COG with this lower cranial radiation dose will be of interest in the years to come, hopefully seeing a reduction in late toxicity without compromising efficacy as cranial RT continues as used in a subset of high-risk pediatric ALL patients.
Meningeal Leukemia at Diagnosis
At the time of diagnosis of ALL, approximately 3% to 5% of patients will present with clinically detectable CNS involvement (i.e., CNS-3 disease). Meningeal leukemia at diagnosis is typically managed as high-risk leukemia with cranial RT. The St. Jude investigators may disagree with the use of RT even here, but their latest results of their Total XV protocol included nine patients with CNS-3 disease whose 5-year event-free survival was only 43%.51 These high-risk patients receive chemotherapy programs that include dose-intensive therapy with agents that penetrate the blood–brain barrier as well as IT therapy. Cranial radiation doses may vary from 18 to 25 Gy. Historically, CSI has been used, but modern practice has dispensed with the spine fields as the IT and systemic chemotherapy programs appear to be effective in assisting with CNS control. In children with ALL, meningeal involvement no longer carries a dire prognosis, as 5-year disease-free survival rates approaching 70% are commonly seen.94
Within the older Children’s Cancer Study Group (CCG) clinical trials for ALL, CSI has been consistently used in the management of CNS-3 disease. Until 1983, the cranial and spinal doses were 24 Gy and 12 Gy, respectively. Subsequent trials through 1989, utilized more intensive consolidation chemotherapy with a decrease in spinal radiation doses to 6 Gy. This allowed for a nonrandomized comparison of 6- and 12-Gy doses to the spine (in 2-Gy fractions). Interestingly, the patients who received a reduced spinal dose did just as well as those who received 12 Gy with less intensive chemotherapy. The 5-year event-free survival rate for patients with CNS-3 disease was 69%, compared to 67% for patients enrolled in all CCG ALL protocols in 1983–1989 who were without CNS-3 disease.94
Delay in RT up to 12 months has been found to be safe as long as intensive chemotherapy is being given first.95,96 This avoids the marrow compromise that could potentially occur with early spine irradiation. In addition, with doses <16 Gy to the spine, myelosuppression has not been a major problem. Musculoskeletal hypoplasia would not be expected to be a significant problem for long-term survivors. The sequencing of RT after rather than before potentially neurotoxic drug therapy such as MTX may theoretically result in a lower incidence of cognitive dysfunction or encephalopathy. Some pediatric protocols will also tailor the dose to the brain based on patient age. For instance, the ALL-BFM 90 protocol, which utilizes cranial rather than CSI, avoids any RT for those younger than 1 year of age, 18 Gy for ages 1 to 2 years, and 24 Gy for older patients. In this large multicenter trial, 54 patients presented with CNS-3 disease and achieved a 48% 6-year event-free survival.58 Arguably, this is inferior to protocols that use CSI, such as those reported by the CCG.94 However, in the ALL-BFM 95, there were 64 patients with CNS-3 disease who had a 6-year event-free survival of 57.7% in which therapy included cranial radiation to 18 Gy without irradiation of the spine.
Summary of Cranial RT for Initial ALL Management
In summary, cranial RT may be used to prevent CNS relapse of leukemia. From a historical perspective that considered the brain as a sanctuary site, cranial RT has been well documented to improve outcomes for selected patients with pediatric ALL. With concerns about late sequela and improvements in systemic therapy including agents that can better penetrate the blood–brain barrier, cranial RT is used much less frequently in the upfront management of ALL. Overall, only 15% to 20% of pediatric ALL patients who have high-risk features require cranial RT currently. In current practice, cranial radiation is employed selectively in high-risk ALL patients. Although the definition of high risk has been shifting, one needs to take into account the intensity and specifics of risk-adapted chemotherapy. Nevertheless, ALL patients who benefit from cranial RT include older age (e.g., older than 10 years of age, and probably increasing benefit at ages older than 13 years); T-cell phenotype, especially in older patients with high presenting WBC over 100.000; and risk groups CNS-2 or -3 by CSF findings. Additionally, among B-cell patients, those with MRD and adverse cytogenetics should be strongly considered for cranial RT. All adults with ALL may be considered to be at high risk for CNS relapse, but there are no standard recommendations on cranial RT. Patients with AML, except for specific subtypes such as monocytic variants, are not generally treated with prophylactic cranial radiation at the present as the risk for CNS relapse is <5%. Unless otherwise indicated, in a specific treatment protocol, the radiation prescription for prophylactic cranial radiation to prevent ALL relapse is 18 Gy in 9 or 10 fractions. When BFM-type chemotherapy programs are used, the radiation dose has been reduced to 12 Gy with favorable outcomes. The spinal region is specifically treated by IT chemotherapy rather than RT, although an open question is whether there would be an incremental benefit to low radiation doses to the spine with meningeal leukemia at diagnosis.
Therapeutic CNS Irradiation for Meningeal Relapse
With modern chemotherapy programs incorporating CNS-directed treatment, CNS relapse rates are typically <10%. As in overt CNS leukemia at diagnosis, RT has a central role. Formally, CNS relapse was thought to have a poor prognosis. Studies in the 1970s and 1980s describe disease control rates of 25% to 50%.97–103 More recent trials, however, utilizing more intensive chemotherapy as well as RT, have reported ALL 5-year survivals of 50% to 70%.95,104–107 Almost all trials have employed RT; the debate has been between cranial RT alone or CSI. Doses used have generally been approximately 24 Gy to the brain and 10 to 15 Gy to the spine. Most comparisons have not been randomized, but superior outcomes seem to be achieved with CSI.58,95,104–105,106,107 One small phase III trial did show superiority for CSI compared to cranial RT.104
Although CNS relapse may ostensibly occur without over systemic disease, the latter is viewed as inevitable without additional systemic therapy. Therefore, intensive chemotherapy is an essential component of the treatment of meningeal relapse.108
Several prognostic factors have been found to be of importance in the setting of CNS relapse.109–111 Patients who were originally deemed at diagnosis to be at low risk for CNS relapse by virtue of a low initial leukocyte count (<20,000/μL), who originally did not receive cranial irradiation, or whose CNS relapse occurred at a relatively long period after the original diagnosis have a better prognosis after CNS recurrence. An isolated CNS relapse has generally been more prognostically favorable compared to combined CNS and systemic relapse. After completing chemotherapy, those children with CNS relapse have better outcomes with longer disease-free intervals. For instance, experience within the Pediatric Oncology Group (POG) with isolated CNS relapse of ALL in which RT used a cranial dose of 24 Gy and a spine dose of 15 Gy. The 4-year event-free survival was 71%. Those patients who presented with greater than an 18-month disease-free interval prior to CNS relapse had a 4-year event-free survival of 83% compared to 46% for those with shorter initial remission durations.95 This has led subsequent POG trials that omit RT to the spine if there is a long disease-free interval, but if the disease-free interval is <18 months, 24 Gy is given to brain while 15 Gy is delivered to the spine. Whether the omission of spinal RT in this favorable subgroup is detrimental is unclear. However, high-dose Ara-C has generally been added to the management of CNS relapse of ALL. With this agent, Mora et al.112 reported a favorable 63% complete response rate. These concepts have been maintained with current COG trials for CNS relapse of ALL, but with intensification of systemic therapy, spinal RT has been proposed to be dropped even for short disease-free intervals, while the brain is treated to 18 Gy. As for adults with CNS leukemia, most of the published experience and commentary from medical oncologists has been opposed to the use of CSI mainly for fear of excessive myelosuppression.87 Cranial irradiation is relatively standard, however. Quite possibly, the avoidance in treating the entire CNS is detrimental, although admittedly this is not a settled issue.
Selected high-risk patients with CNS relapse may be candidates for allogeneic transplants. TBI is often a component of the preparative regimen. One function of the TBI is to specifically treat the CNS burden of disease. Because doses on the order of 12 to 15 Gy are employed here, it makes sense to boost the head prior to TBI to bring total doses to the cranium to 18 to 25 Gy.
Testicular Relapse
In boys with ALL, particularly those with T-cell subtype, testicular involvement was once a common problem. Overt testicular involvement by leukemia at diagnosis occurs in approximately 2% of males with ALL, and is a particularly poor prognostic situation. Microscopic burden in the testes is estimated to be higher at 5% to 15% based on biopsy data and the historic risk for testicular relapse. With modern chemotherapy, especially the use of intermediate- to high-dose MTX, this is now rare. Similar to the experience with CNS relapse, there once was speculation as to whether the testes acted as a sanctuary site due to a physiologic blood–testes barrier. In 1980, a trial of prophylactic testicular irradiation significantly reduced the risk of testicular relapse but did not improve survival.113
In cases of testicular relapse, systemic and/or CNS relapse usually follows.114 Both intensive systemic therapy and local RT are indicated. Doses <12 Gy are generally thought to be suboptimal, whereas doses of 24 to 26 Gy over 2.5 to 3.5 weeks are considered standard. Case reports of local recurrences despite adequate RT have led some to suggest higher doses. When only one testes is clinically involved, imaging or biopsy of the contralateral testes frequently reveals bilateral disease. Similarly, unilateral irradiation or orchiectomy as local management is felt to be associated with a significant risk of contralateral testicular relapse, justifying treatment directed at both testes for leukemia management, despite the expectation of infertility from RT. Data from CCG and POG studies suggests that local irradiation and intensive systemic therapy results in prolonged event-free survival in roughly 50% to 65% of patients.61,115 Where allogeneic transplantation is indicated, TBI is often part of the conditioning regimen. One retrospective series from the Memorial Sloan-Kettering Cancer Center suggests that for TBI/cyclophosphamide preparative regimens, the risk of testicular relapse is significantly reduced with a local boost to the scrotum of 4 Gy to bring the total testicular dose to 16 to 20 Gy.116 Regardless, there is not universal agreement about the need for a testicular boost in this setting.
 THE CHRONIC LEUKEMIAS
THE CHRONIC LEUKEMIAS
Chronic Myelogenous Leukemia
CML is a chronic myeloproliferative disorder arising from clonal expansion of the primitive hematopoietic stem cell. It involves myeloid, erythroid, megakaryocytic, and sometimes lymphoid elements. It is the first neoplastic process to be characterized by a specific cytogenetic marker, the Philadelphia chromosome (Ph+), t(9;22) described in 1960.117 This is detectable by cytogenetics in 90% to 95% of patients and by molecular analysis in most other patients.118 This translocation results in a fusion protein that has tyrosine kinase activity critical for leukemic transformation.
Clinically, the disease manifests several phases: an initial chronic indolent phase of 3 to 4 years with progression to an accelerated phase and finally an acute transformation to blast phase, which occurs in 75% to 85% of patients, with a survival of 3 to 6 months. As the disease advances, the accelerated phase is characterized by increasing difficulty in controlling the peripheral WBC count, increasing splenomegaly, increasing blasts in the peripheral blood and bone marrow, and increasing basophilia and eosinophilia. The blast crisis resembles acute leukemia with >30% blasts in blood or bone marrow with symptoms such as bone pain, sweats, fever, anorexia, or weight loss. Anemia, thrombopenia, and extramedullary disease involving bones, skin, CNS, and lymph nodes are common. In about 20% of cases, blasts are lymphoid by phenotype.
Prognostic factors have been identified in CML and include age, splenic size, platelet count, percent of basophils in blood, and marrow. Poor prognosis factors are age >60 years, spleen >l0 cm below the costal margin, blasts >3% in blood or marrow, basophilia >7% in blood or marrow, and platelets >700,000/μL.119,120 A poor response to therapy and a short duration of remission also are considered unfavorable, as well as failure to achieve significant cytogenetic remission.121
Therapy of CML
The first effective therapy for chronic leukemias was RT to the spleen and sometimes the liver, initiated in 1902 by Pusey122 and assessed in 1924 by Minot et al.123 Today, RT is primarily used in a palliative setting to relieve painful splenomegaly or other extramedullary sites when indicated. In some centers, TBI plays an important role in allogeneic transplantation.
There is a long history of treating CML in chronic phase with alkylating agents, hydroxyurea, cytarabine,and interferon alpha, which is beyond the scope of this chapter. Over the past decade, such therapies, as well as hematopoietic stem cell transplant, have largely been relegated to the salvage setting through the introduction of ABL tyrosine kinase inhibitors effective in inducing and maintaining long-term remissions in CML.124 Imatinib is a relatively nontoxic oral medication that has been shown to be effective in CML in both chronic and accelerated phases.125,126 This agent has been a paradigm for molecularly targeted therapies and is now used as upfront therapy for CML along with two other more recently introduced agents dasatinib and nilotinib.127 For those who do not respond adequately to the tyrosine kinase inhibitors or who lose response, allogeneic transplantation remains an important option.128,129 About 70% of good-risk patients achieve long-term disease-free survival.130 Allotransplantation and total body irradiation are further discussed in Chapter 15.
Palliative RT may be of benefit in those patients with massive splenomegaly with responses seen with doses in the 10- to 20-Gy range or lower. The radiation oncologist is well advised to understand the extreme radiosensitivity of the malignant stem cells in CML and in other hematopoietic disorders causing splenomegaly with the admonition to use very low doses per fraction (25 to 100 cGy) once or twice a week with close monitoring of blood counts (see below).
TABLE 80.3 CHRONIC LYMPHOCYTIC LEUKEMIA STAGING SYSTEMS