ISOLATED HYPOALDOSTERONISM
Part of “CHAPTER 81 – HYPOALDOSTERONISM“
Isolated hypoaldosteronism, a selective deficiency of aldosterone secretion without alteration in cortisol production, results in a persistent hyperkalemia, which may be associated with profound muscle weakness and cardiac arrhythmias. Isolated hypoaldosteronism can result from inborn errors in aldosterone biosynthesis, failure of the zona glomerulosa owing to autoimmune adrenal disease in association with critical illness, altered function of the renin-angiotensin system in the syndrome of hyporeninemic hypoaldosteronism, unilateral adrenalectomy for an aldosterone-producing adenoma, or pharmacologic inhibition of aldosterone. Mineralocorticoid resistance occurs when there is a lack of response to aldosterone despite the presence of this hormone.1
PRIMARY DEFICIENCY
INBORN ERRORS
Inborn errors in the oxidation of corticosterone to form aldosterone have been described as corticosterone methyl oxidase type I deficiency (CMO I) and corticosterone methyl oxidase type II deficiency (CMO II). Corticosterone is at first hydroxylated and oxidized at the 18th position to yield aldosterone. This sequence of enzymatic events is seen in Figure 81-1. CMO I is also referred to as 18-hydroxylase; CMO II is also referred to as aldosterone synthase or aldosterone oxidase. CMO II deficiency is more common than CMO I deficiency. Some of the CMO I and CMO II enzymes have activity residing in the isozyme of steroid 11β-hydroxylase; this isozyme activity is limited to the zona glomerulosa. 18-Hydroxylase and 18-oxidase activities are required for the production of aldosterone. Mutations in the genes that encode the isozyme result in defective synthesis of aldosterone.
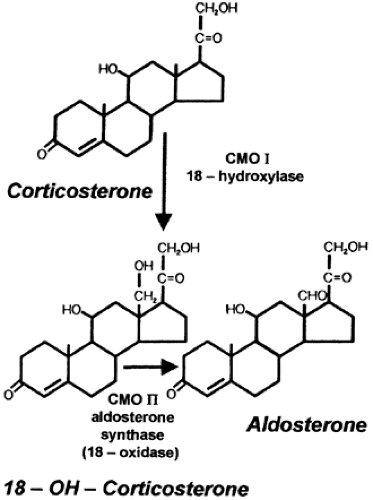 FIGURE 81-1. Terminal steps in the biosynthesis of aldosterone. Corticosterone methyl oxidase (CMO) type I and II deficiencies exhibit defects in the hydroxylase-oxidase reactions. In CMO type I deficiency, levels of the 18-hydroxylated product, 18-OH-corticosterone, are reduced. In CMO type II deficiency, 18-OH-corticosterone production is markedly increased. In both cases, aldosterone deficiency results. |
Corticosterone Methyl Oxidase Type I Deficiency (18-Hydroxylase Deficiency).
CMO I deficiency is extremely rare. Biochemically, it is characterized by a marked overproduction of corticosterone by the zona glomerulosa without a corresponding increase in 18-hydroxycorticosterone and a virtual absence of aldosterone. Scrutiny of some case reports casts doubt on whether they represent type I or type II deficiency because 18-hydroxycorticosterone measurement was not available. However, clinical results for a North American kindred in Pennsylvania, although attributed to CMO II deficiency, probably represent the type I variant.2,3
Corticosterone Methyl Oxidase Type II Deficiency (Aldosterone Synthase Deficiency).
CMO II deficiency is inherited as an autosomal-recessive trait. This deficiency is rare but has been observed with an increased frequency among Jews of Iranian origin. The biomolecular studies have been extensive.4
In both CMO I and CMO II deficiencies, the severity of the clinical manifestations is inversely related to the age at diagnosis: it becomes less severe as the child ages. CMO II deficiency, if recognized clinically, has its onset between 1 week and 3 months of age and is characterized by severe dehydration, vomiting, and failure to grow. Hyponatremia, hyperkalemia, and metabolic acidosis are uniformly present. The plasma renin activity (PRA) is elevated, and plasma aldosterone levels are low. On the other hand, plasma 18-hydroxycorticosterone levels are markedly increased, and the 18-hydroxycorticosterone/aldosterone ratio in plasma exceeds 5. Also, the ratio of the urinary metabolite of 18-hydroxycorticosterone, 18-hydroxytetrahydroaldosterone, to the metabolite of aldosterone, tetrahydroaldosterone, also exceeds 5. In older children, adolescents, and adults, the abnormal steroid pattern described may be present and may persist throughout life without clinical manifestations.
Mineralocorticoid (fludrocortisone) is given during infancy and early childhood, but this therapy does not have to be continued in most cases. Moreover, spontaneous normalization of growth can occur in patients who are untreated. It is not understood why aldosterone deficiency is so much more threatening in infancy than in adult life. It is particularly puzzling that isolated hypoaldosteronism caused by low renin secretion in aging patients has clinical significance, whereas patients with asymptomatic inherited hypoaldosteronism, resulting from either CMO I or CMO II deficiency, exhibit none of the manifestations of hyporeninemic hypoaldosteronism. These same observations are true of patients with pseudohypoaldosteronism (PHA).
FAILURE OF ADRENAL GLOMERULOSA FUNCTION
Autoimmume Adrenal Failure.
As autoimmune adrenal failure evolves, selective aldosterone deficiency may emerge in the presence of preservation of zona fasciculata cell function. Although glucocorticoid responsiveness to corticotropin (ACTH), metyrapone, or insulin-induced hypoglycemia may be normal, PRA is elevated in the presence of a low or undetectable plasma corticotropin level. This is accompanied by mild metabolic acidosis and, occasionally, hyponatremia. During late stages of the disease, progression to panadrenal insufficiency may occur. A period of up to 1 year may separate the onset of the mineralocorticoid and glucocorticoid deficiencies.5
In selective aldosterone deficiency caused by autoimmune disease, antiadrenal antibodies can be detected. Mucocutaneous candidiasis and hypoparathyroidism may occur concurrently,6 a form of multiple autoimmune endocrinopathy.
A patient with idiopathic hemochromatosis, weakness, postural hypotension, and loss of libido was found to have mild glucose intolerance and low gonadotropins in addition to normokalemia, hyponatremia, and a modest elevation of urea. PRA was elevated, and aldosterone levels were suppressed. Cortisol response to ACTH and urinary 17-hydroxysteroid were normal. The patient failed to conserve sodium on a sodium-restricted diet. This suggests that isolated mineralocorticoid deficiency can occur secondary to glomerulosa cell failure caused by iron deposition.7
Hypoaldosteronism in Ill Patients.
Hyperreninemic hypoaldosteronism can occur in critically ill patients, such as those in septic states, or in hemodynamically compromised subjects. Most of these patients have prolonged illness and hypotension of long duration, with or without hyperkalemia. The cortisol secretion is elevated, commensurate with the level of the stressful state. Because aldosterone, corticosterone, and 18-hydroxycorticosterone (but not cortisol) secretions become suppressed within 48 to 96 hours of continuous ACTH stimulation of the adrenal gland,8 prolonged ACTH secretion secondary to stress may impair 11β-hydroxylase and 18β-hydroxylase enzymes and may be the underlying mechanism of this syndrome. However, these patients have an increased plasma 18-hydroxycorticosterone/aldosterone ratio, and their aldosterone response to angiotensin II (A-II) infusion is impaired; this suggests that a selective inhibition of CMO II may play a role in the development of this disease. Because hypoxia is associated with increased renin activity and cortisol secretion and diminished aldosterone secretion,9 insufficiency of CMO II activity may be the result of hypoxia or other circulating factors affecting the zona glomerulosa. Autopsy studies in adrenal glands from patients with this syndrome have revealed atrophy or necrosis involving most of the layers of the adrenal gland, including the zona glomerulosa. However, an isolated necrosis of the zona glomerulosa has not been found; this is probably because blood flows from the outer cortex toward the medulla, and hypoper-fusion would affect the inner corticomedullary region first.10
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


