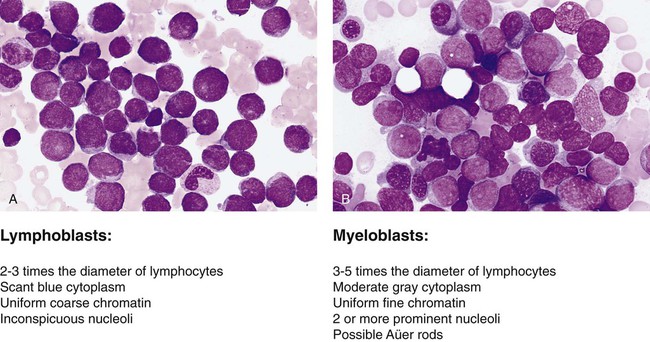
After completion of this chapter, the reader will be able to: 1. Name two viruses that play a pathogenic role in lymphoid malignancies. 2. Define oncogene and tumor suppressor gene and explain the difference between them, including the mechanisms by which each produces cancer. 3. Explain why oncogenes are described as acting in a dominant fashion. 4. Give two examples of oncogenes and two examples of tumor suppressor genes. 5. Describe how genetic abnormalities can affect molecular pathways within cells. 6. Identify the two basic subgroups of chemotherapeutic agents. 7. Explain why localized treatments are rarely used for leukocyte neoplasms. 8. Describe examples of supportive care therapies that have been developed from molecular biology techniques, including their functions and purposes. 9. Name two examples of targeted therapeutics for chronic myelogenous leukemia and explain their methods of action. 10. Describe the differences between small molecule inhibitors and monoclonal antibodies. 11. Name three sources of hematopoietic stem cells. 12. Compare and contrast allogeneic and autologous bone marrow transplants. The French-American-British (FAB) classification of the acute leukemias was devised in the 1970s and 1980s. The FAB schemas were based largely on morphologic characteristics and relied heavily on examination of routine histologic stain preparations to distinguish lymphoid neoplasms from myeloid neoplasms (Figure 29-1). Although these types of diagnostic criteria have not been abandoned, pathologists are now moving toward more precise classification of many of the leukocyte neoplasms based on recurring chromosomal and genetic lesions found in many patients. These lesions are related to disruptions of oncogenes, tumor suppressor genes, and other regulatory elements that control proliferation, maturation, apoptosis, and other vital cell functions. In 2001 the World Health Organization (WHO) published new classification schemes for nearly all of the tumors of hematopoietic and lymphoid tissues.1 In some cases WHO melded the older morphologic schemes with the newer schemes. For instance, in the WHO classification scheme for acute myeloid leukemias (AMLs) there are some remnants of the old FAB classification, but new classifications were introduced for leukemias associated with consistently recurring chromosomal translocations. This 2001 WHO classification of hematologic malignancies has undergone a recent revision.2 In part because of the ease of access to bone marrow compared with other organs of the body, researchers have learned a great deal about the biology of cancer by studying hematopoietic neoplasms. The study of chromosomal translocations in hematologic malignancies has taught how a single mutation, or series of mutations in stepwise fashion, can lead to malignant transformation by disrupting the molecular machinery of the cell. The first two genetic lesions found in any kind of human cancer were identified as chromosomal translocations occurring in leukocyte malignancies. These were the t(9;22) translocation in chronic myelogenous leukemia (CML)3 and the t(8;14) translocation in Burkitt lymphoma.4 Tumor suppressor genes are so named because they code for proteins that resist malignant transformation. These genes do not act in a dominant fashion as in the case of oncogenes; rather, cells are transformed into a malignant phenotype only after both alleles of these genes have been lost or otherwise inactivated, the so-called two-hit mechanism proposed by Knudson.5 Although tumor suppressor genes are harder to isolate and identify, numerous such genes have now been identified, and many have been found to be associated with autosomal dominant familial cancer predisposition syndromes. Some well-known examples include the RB1 tumor suppressor gene involved in familial retinoblastoma, the TP53 gene in Li-Fraumeni syndrome, the WT1 gene in Wilms tumor, and the NF1 gene in familial neurofibromatosis type 1. Perhaps more importantly, many of these tumor suppressor genes and their protein products are altered in many sporadic cancers, including hematologic cancers. The list of chromosomal and molecular aberrations known to occur in the various leukocyte neoplasms continues to grow on an almost daily basis. Indexing this list is far beyond the scope of this chapter, but some condensed lists are provided in Chapter 31. More complete indices can be found in other publications such as the WHO reclassification scheme,1 its 2008 revision, or other hematology textbooks.6–10
Introduction to Leukocyte Neoplasms
Classification Schemes for Leukocyte Neoplasms
Chromosomal Abnormalities in Hematologic Neoplasms
Tumor Suppressor Genes
Molecular Pathways Perturbed by Cellular Transformation
Oncohema Key
Fastest Oncology & Hematology Insight Engine