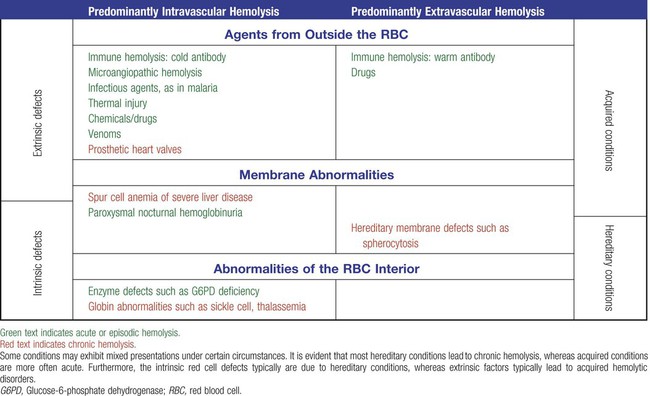
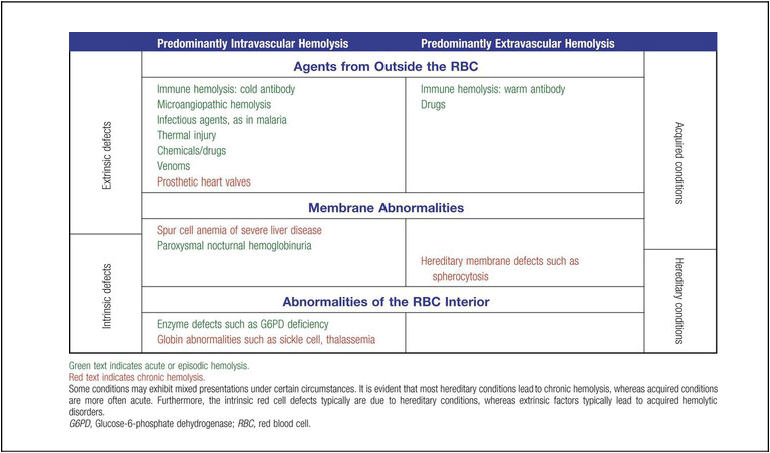
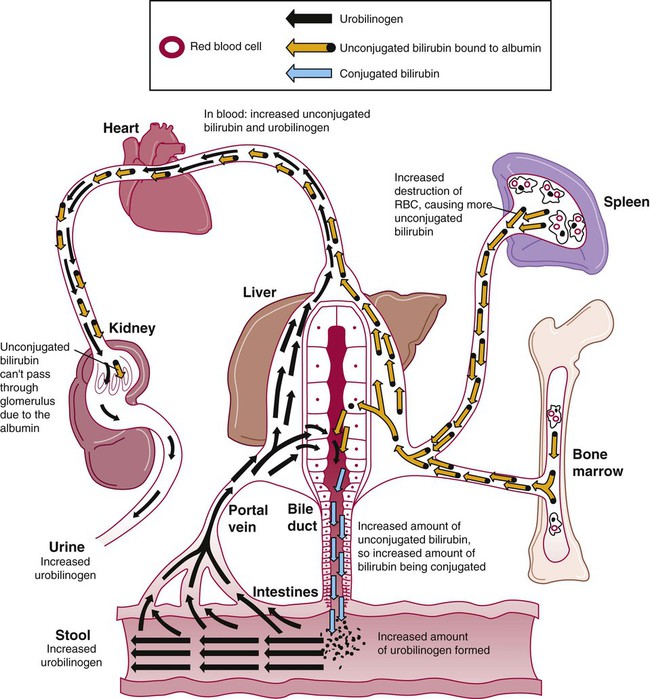
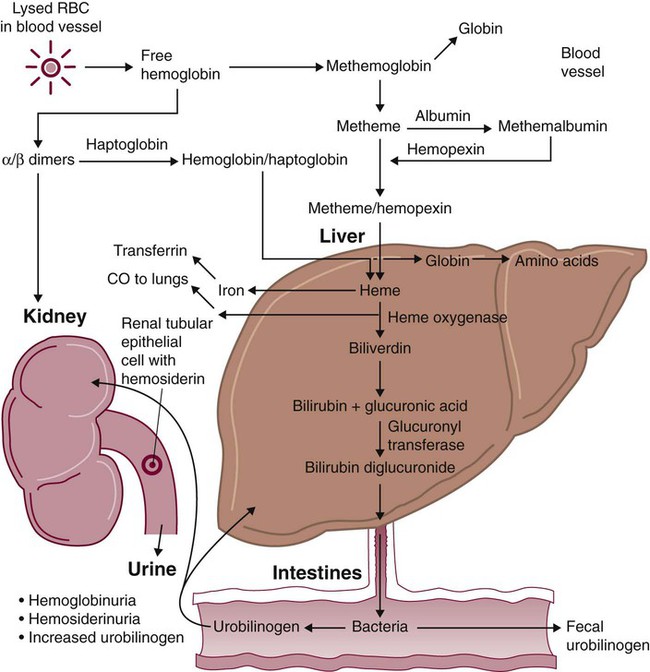
After completion of this chapter, the reader will be able to: 1. Define hemolysis and recognize its hallmark clinical findings. 2. Differentiate a hemolytic disorder from hemolytic anemia by definition and recognition of laboratory findings. 3. Discuss methods of classifying hemolytic anemias and apply the classification to an unfamiliar anemia. 4. Describe the processes of intravascular (fragmentation) and extravascular (macrophage-mediated) hemolysis, emphasizing sites of hemolysis for each and associating the clinical findings of each. 5. Differentiate intravascular and extravascular hemolysis when given clinical and laboratory findings. 6. Describe the process of normal red blood cell (RBC)/hemoglobin catabolism, emphasizing the process for catabolism of protoporphyrin. 7. Identify laboratory tests that indicate accelerated RBC destruction, explain the physiologic mechanisms involved, and interpret the results of each test. 8. Identify, explain, and interpret the results of laboratory tests that indicate increased erythropoiesis. 9. Differentiate between hemolytic anemias and other causes of increased erythropoiesis given laboratory or clinical information. 10. Describe the mechanisms that salvage hemoglobin during intravascular hemolysis. 11. Explain the rationale for tests of the hemoglobin salvage mechanisms and interpret the results of each. 1. What process is indicated by the root beer–colored urine? 2. What laboratory tests can be used to differentiate the cause of the hemolysis? 3. Based on the patient’s clinical presentation, predict the results expected for each test listed for question 2. This chapter presents an overview of the hemolytic process and provides a foundation that is applicable in the following chapters on red blood cell (RBC) disorders. The term hemolysis or hemolytic disorder refers to increased destruction (i.e., lysis) of RBCs, shortening their life span. The reduced number of cells results in reduced tissue oxygenation and increased erythropoietin production by the kidney. When the patient is otherwise healthy, the bone marrow responds by accelerating erythrocyte production, which leads to reticulocytosis. A hemolytic process is present without anemia if the bone marrow is able to compensate by accelerating RBC production sufficiently to replace the RBCs lost through hemolysis. Healthy bone marrow can increase its production of RBCs by six to eight times normal1; therefore, significant RBC destruction must occur before an anemia develops. A hemolytic anemia results when the rate of RBC destruction exceeds the increased rate of RBC production. When hemolysis is the primary feature, anemias can be classified as follows: Every hemolytic condition can be classified according to each of these descriptors. Table 22-1 shows this and provides a noncomprehensive list of hemolytic anemias. This chapter focuses on the distinction between intravascular and extravascular hemolytic conditions. The other classifying schemes are summarized here briefly and are used to organize the chapters that follow. Extrinsic hemolytic conditions are those that arise from outside the RBC, typically substances in the plasma or conditions affecting the anatomy of the circulatory system. Even though malaria protozoa and other infectious agents are within the RBC, they are classified as extrinsic because the RBC was normal until it was invaded by an outside agent. An antibody against RBC antigens and a prosthetic heart valve are examples of noninfectious extrinsic agents. In extrinsic hemolysis, cross-transfusion studies have shown that the patient’s RBCs have a normal life span in the bloodstream of a normal individual, but normal cells are lysed more rapidly in the patient’s circulation. These studies confirm that something outside the RBCs is causing the hemolysis. (Of course, in the case of intracellular parasites, the cross-transfusion study is not applicable.) Most intrinsic defects are inherited; most extrinsic ones are acquired (see Table 22-1). A few exceptions exist, such as paroxysmal nocturnal hemoglobinuria, an acquired disorder involving an intrinsic defect (see Chapter 23). Intrinsic disorders are subclassified as membrane defects, enzyme defects, and hemoglobinopathies. Extrinsic hemolysis may be immunohemolytic, traumatic, or microangiopathic, or may be caused by infectious agents, chemical agents (drugs and venoms), or physical agents (see Table 22-1). RBCs live approximately 120 days. During this time, they undergo various metabolic and chemical changes, which result in a loss of deformability. Under normal circumstances, macrophages of the mononuclear phagocyte system (or reticuloendothelial system) recognize these changes and phagocytize the aged erythrocytes (see Chapter 8). The organs of this system include the spleen, bone marrow, liver, lymph nodes, and circulating monocytes, but it is primarily the spleen and liver that process senescent RBCs. The macrophages in the spleen (littoral cells) are especially sensitive to subtle RBC changes, whereas the macrophages of the liver (Kupffer cells) detect and destroy more severely damaged RBCs. This is an extravascular hemolytic process. Hemolysis occurs within macrophages; thus macrophage-mediated hemolysis is a more precise term. The majority of RBC degradation occurs extravascularly as enzymes of the macrophage lyse the phagocytized erythrocytes (Figure 22-1). Hemoglobin is hydrolyzed into heme and globin; the latter is further degraded into amino acids that return to the amino acid pool. Iron is released from the heme, returned to the plasma via ferroportin, bound to its protein carrier molecule (transferrin), and recycled to developing RBCs. The remaining protoporphyrin is degraded through a series of biochemical reactions in different tissues and organs. Figure 22-1 illustrates protoporphyrin catabolism. While protoporphyrin is inside the macrophage, heme oxygenase acts on it, breaking the porphyrin ring to yield a linear molecule, biliverdin. The lungs excrete a by-product of that reaction, carbon monoxide. The green biliverdin is reduced to bilirubin, a nonpolar yellow molecule that is secreted into the plasma (Box 22-1). Because it is hydrophobic, this bilirubin must bind to albumin to be transported in plasma to the liver. The bilirubin-albumin complex enters the liver parenchymal cells, where the bilirubin dissociates from the albumin and is joined (i.e., conjugated) with two molecules of glucuronic acid by glucuronyl transferase to form bilirubin diglucuronide.2 The addition of the two sugar acid molecules makes the molecule polar and water soluble. Bilirubin diglucuronide is also called conjugated bilirubin. The bilirubin originally released from macrophages that lacks these sugars is termed unconjugated. Intravascular hemolysis is trauma to the RBC membrane that causes a breach sufficient for the cell contents, chiefly hemoglobin, to spill directly into plasma (Box 22-2). Approximately 10% to 20% of normal RBC destruction is intravascular,3 secondary to turbulence and anatomic restrictions in the vasculature. Because hemoglobin is filtered by the kidney, iron could be lost in normal intravascular hemolysis. In addition, free hemoglobin and iron can cause oxidative damage to cells. Several mechanisms exist to salvage hemoglobin iron and prevent oxidation reactions, and are collectively called the haptoglobin-hemopexin-methemalbumin system (Figure 22-2). When free in the plasma, hemoglobin exists as α/β dimers bound to a liver-produced plasma protein called haptoglobin. This is the first mechanism of hemoglobin iron salvage. By binding to haptoglobin, hemoglobin avoids filtration at the glomerulus and is saved from urinary loss. The complex is carried to the liver, where the haptoglobin-hemoglobin complex is bound to macrophage receptors and internalized into the macrophage.4 Inside the macrophage, iron is salvaged, and the remaining protoporphyrin is converted to bilirubin, just as though the RBC had been ingested by the macrophage. If lysis is accelerated, haptoglobin is depleted, because the liver’s production of haptoglobin does not increase in response to the increased hemolysis and is typically adequate to salvage only a normal amount of plasma hemoglobin. A secondary mechanism of iron salvage involves hemopexin. In hemoglobin that is not bound by haptoglobin, the iron becomes oxidized, forming methemoglobin. The heme molecule (actually metheme) dissociates from the globin and binds to another liver-produced plasma protein, hemopexin.5,6 This binding also saves the iron from urinary loss and prevents oxidation of cell membranes. Hemopexin-metheme binds to hepatocyte receptors and is internalized. There the iron is salvaged, and the protoporphyrin is converted to bilirubin, as in a macrophage. In contrast to haptoglobin, hemopexin is recycled to the plasma from the hepatocyte, so a single hemopexin molecule collects and salvages more free metheme molecules. Although its production is constant, because it is recycled to the plasma, levels of hemopexin do not fall dramatically during periods when there is an increase in intravascular hemolysis. A third mechanism of iron salvage is the metheme-albumin system. Albumin acts a carrier for many molecules. Metheme can bind to albumin when hemopexin is saturated, which further reduces urinary loss. This is probably a temporary holding state for the metheme, which transfers to hemopexin because it has a higher binding affinity for metheme than does albumin.7 It is then transported to the liver for processing. If the previous systems are overloaded, the excess hemoglobin or heme iron will be filtered into the urine. Iron still can be retained in the kidney, however.8,9 Some hemoglobin or (met)heme that enters the filtrate is reabsorbed, not by specific carriers but as part of the general filtrate in the proximal tubule. When inside the renal tubular cell, the iron is separated from the protoporphyrin. There it is stored as ferritin or hemosiderin. The role of the kidney in salvaging iron and returning it to the circulation is uncertain at this time. Although carrier proteins like divalent metal transporter 1 and ferroportin have been identified in the kidney of experimental mice, their localization, function, and regulation are still not clear. When the amount of hemoglobin presented to the kidney exceeds what can be reabsorbed, the excess is then detectable in the urine. Many hemolytic anemias are a result of increased extravascular hemolysis (Figure 22-3) during which more than the usual number of RBCs are removed from the circulation daily. Under normal circumstances, senescent RBCs display surface markers that identify them to macrophages as aged cells requiring removal (see Chapter 8). Pathologic processes also lead to expression of abnormal markers, so that cells are identified and removed. If the number of affected cells increases beyond the quantity normally removed each day due to senescence, and if the bone marrow cannot compensate, then anemia develops. As an example, Heinz bodies, aggregates of denatured hemoglobin formed in various anemias, bind to the inner surface of the RBC membrane, producing changes to the exterior of the membrane that can be detected by macrophages. When something causes increased formation of Heinz bodies or other intracellular inclusions, the cells are removed from the circulation prematurely by macrophages. A similar process occurs when intracellular parasites are present or when complement or immunoglobulins are on the surface of the RBC.
Introduction to Increased Destruction of Erythrocytes
Case Study
Classification
Hemolysis
Normal Bilirubin Metabolism
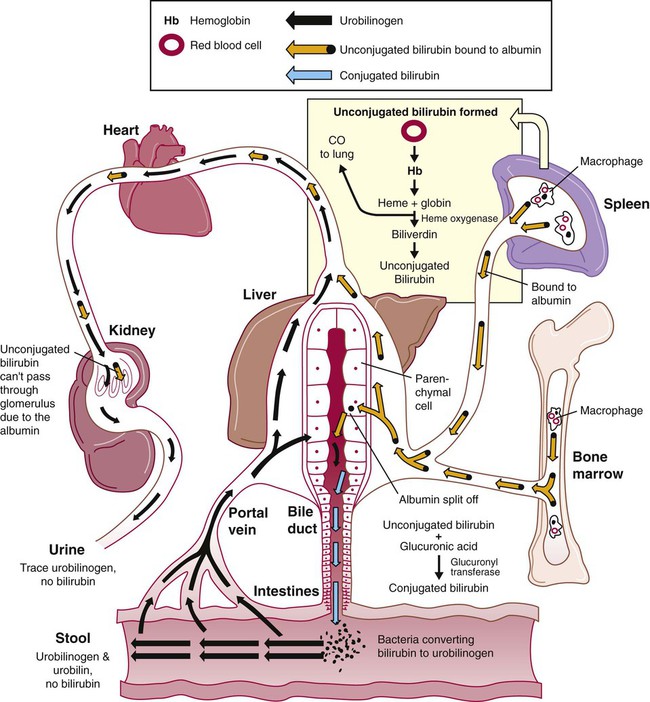
Normal Plasma Hemoglobin Salvage During Intravascular Hemolysis
Excessive Extravascular Hemolysis
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Oncohema Key
Fastest Oncology & Hematology Insight Engine