Classification of diabetes
Type 1 diabetes (T1D)
Type 2 diabetes (T2D)
Gestational diabetes
Synonym
IDDM, juvenile-onset diabetes
NIDDM, adult-onset diabetes
GDM
Epidemiology
5–10 % of total cases
90–95 % of total cases
3–10 % of pregnancies
Risk group
Typically <20-years old
Adults (≥25-years)
Pregnant women >25-years old
Risk factors
Susceptible HLA genes, autoantibodies, environmental factors (dietary factors and viral infection)
Obesity, old age, and unhealthy lifestyle
Placental hormones, elevated BMI before pregnancy, age of conception, and race
Symptoms
Polydypsia, polyphagia, polyuria and weight loss
Polydypsia, polyphagia, polyuria and weight loss, fatigue, slow wound healing
As for T1D and T2D
Etiology
Autoimmune β-cell destruction, idiopathic
Insulin resistance
Insulin resistance or deficiency
Diagnosis
HbA1c level >6.5 %, GAD, insulin and IA2 antibodies and HLA genotyping
Fasting BGL ≥7 mmol/L, 2 h BGL ≥7.8 mmol/L
OGTT (75 g glucose load, fasting BGL ≥7 mmol/L, 2 h BGL ≥7.8 mmol/L)
Complications
Micro- and macrovascular diseases. Prominence of microvascular (e.g., retinopathy, nephropathy, and neuropathy)
Micro- and macrovascular diseases. Prominence of macrovascular (e.g., heart disease, hyperlipidemia and ketoacidosis)
Congenital defects, neonatal hypo/hyperglycemia, fetal macrosomia, and perinatal mortality
Insulin, an endogenous hormone is secreted by b-cells present in the Islets of Langerhans of the pancreas and is responsible for maintaining homeostatic, euglycemic levels (80–110 mg/dL or 4.5–6.2 mmol/L) within the body (Fig. 1.1). Lowered insulin secretion elevates blood glucose levels beyond the normal range, leading to hyperglycemia. Insulin facilitates glucose utilization, energy generation and stores energy, predominantly in the hepatocytes, adipocytes, and myocytes. The hormone is first synthesized in its precursor form which is known as ‘preproinslin’, after which proteolytic enzymes cleave the signal sequence and generate ‘proinsulin’ (86 amino acid containing protein) arranged in three distinct peptide chains: A- (21 amino acid), C- (31 amino acid), and B-chain (30 amino acid). The protein is further processed by proteases within endoplasmic reticulum (ER), cleaving the C-peptide (C-chain) and forming a 51 amino acid bridged peptide, which is active insulin. The discarded C-peptide portion of proinsulin is a reliable marker of insulin secretion and is routinely used as a diagnostic marker, as well as to develop therapies and better understand the pathophysiology of diabetes mellitus.
T1D is an autoimmune disease, usually diagnosed in children and young adults, where pancreatic β-cells are attacked by autoantigens, that is, an immune-mediated attack against one’s own tissues, specifically, insulin producing β-cells in the Islets of Langerhans of the pancreas resulting in depleted insulin levels, which, if left untreated, leads to hyperglycemia. There is substantial credible evidence highlighting that autoimmunity is the root cause of T1D, this stemming from the gross infiltration of Islet of Langerhans by antigen presenting cells (APCs) such as macrophages (Mϕ), dendritic cells (DCs) as well as B- and T-lymphocytes (CD4+ and CD8+) (Nerup et al. 1994; Kaufman 2003; Pihoker et al. 2005; Kabelitz et al. 2008).
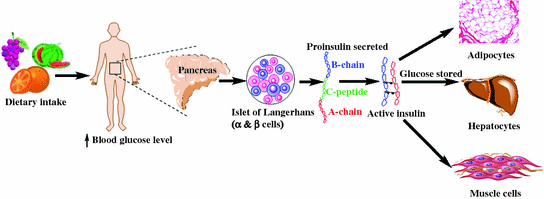
Fig. 1.1
General scheme of insulin biosynthesis and physiological function
The human adaptive immune system possesses a characteristic feature to respond to the foreign/non-self-antigens, wherein it fails to react to antigen derived from self-tissues (self-antigens/autoantigens). However, for unknown reasons, the immune system mistakenly identifies these autoantigens as foreign bodies and mounts an immune response against them. Once these autoantigens are identified, peptide major histocompatibility complex (pMHC) expressed on the surface of APCs (DCs and mTECs; medullary thymic epithelial cells) within thymic medulla complexes with the T-cell receptor (TCR) on the T-cell’s surface (shown in Fig. 2.2) (Concannon et al. 2009). Even after the identification of these autoantigens, a vast population fails to develop autoimmunity due to active self-tolerance mechanisms (Parish and Heath 2008). In a state of active tolerance, autoreactive T-cells developed toward autoantigens are then purged out of the system either by (a) central/thymic tolerance or (b) peripheral tolerance. Central tolerance occurs within the thymus gland wherein T-cells undergo positive and negative selection. In positive selection, T-cells are given survival signal following their binding with MHC/peptide complex with adequate affinity. During development within thymus, T-cells are educated to delete T-cells with high avidity T-cell receptor (TCR) for autoantigens (self-reactive). These self-reactive T-cells are purged either by; (1) clonal deletion, i.e., physiological deletion of the autoreactive T-cells, or (2) anergy, i.e., functional inactivation of the autoreactive T-cells. On the other hand a minor population of low avidity autoreactive T-cells often manages to survive and escape central tolerance processing, migrating into the periphery. These low avidity T-cells in the periphery are of particular concern given their ability to be processed by the host immune system are a danger to the host immune system, with the potential of developing autoimmunity. Although these autoreactive T-cells bypass the central tolerance, they can still be curbed from spreading within the periphery by what is known as peripheral tolerance (Mueller 2009). One of the purported regulatory actions of peripheral tolerance is self–ignorance, whereby autoreactive T-cells persist in the periphery but in a naïve state, where they are incapable of imparting any deleterious autoimmune effects. However, it has been shown in a range of experimental models that, if ‘self’ pMHC complexes are sufficiently outnumbered, the low avidity T-cells may be activated, once again leading to the genesis of autoimmunity (Zehn and Bevan 2006; Henrickson et al. 2008).
Related posts:
 Predictors and Pathogenesis of Type 1 Diabetes
Predictors and Pathogenesis of Type 1 Diabetes
 Triggers Causing Type 1 Diabetes
Triggers Causing Type 1 Diabetes
 Type 1 Diabetes: Past, Present, and Future Therapies
Type 1 Diabetes: Past, Present, and Future Therapies
 Female reproduction: VI Contraception
Female reproduction: VI Contraception
 Psychological Stress, Immunity, and the Effects on Indigenous Microflora
Psychological Stress, Immunity, and the Effects on Indigenous Microflora
 Cardiovascular and Metabolic Syndrome Impact on Uristatin
Cardiovascular and Metabolic Syndrome Impact on Uristatin
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


