Chapter 53 International Parasitic Infections
INTRODUCTION
Some of the common parasitic diseases found in those living with HIV are covered in other chapters, including toxoplasmosis (Chapter 37), cryptosporidia, isospora, cyclospora infections (Chapter 38), and microsporidiosis (Chapter 39). This chapter will focus on those parasitic infections not usually acquired in the United States or Western Europe where differences in epidemiology, clinical manifestations, pathology, therapy, or prognosis in HIV-positive populations have been established. These diseases include Chagas’ disease, leishmaniasis, malaria, and schistosomiasis.
CHAGAS’ DISEASE (TRYPANOSOMA CRUZI INFECTION)
Trypanosoma cruzi, causative agent of Chagas’ disease, is the third most common parasitic infection in humans, after malaria and schistosomiasis. It is found throughout South America and is transmitted by the reduviid bug and less frequently by blood transfusion or organ transplantation.1,2 In the immunocompetent host, primary infection is associated with nonspecific illness. In less than 5% of infected individuals, chronic disease develops in the myocardium, colon, or esophagus, causing cardiomyopathy or a megasyndrome. More commonly, the parasite remains in the host in an indeterminate phase that is clinically undetectable.
Clinical Presentation
During immunosuppression, T. cruzi infection in the indeterminate phase may reactivate as the parasites replicate within the host and cause end-organ disease. In people living with HIV infection, this usually occurs when CD4+ T-lymphocyte counts fall below 200–300 cells/mm3. The most common site of reactivation for HIV-infected patients is the central nervous system (CNS); patients present with a meningoencephalitis syndrome.3–10 Trypanosome invasion of the brain forms chagoma masses, causing patients to present with headache, fever, cognitive changes, and seizures. Focal neurological impairments resulting from chagomas include tremors, hemiparesis, cranial nerve abnormalities, and aphasia.11
After the central nervous system, the second site most commonly affected during reactivation is the heart.12 Cardiac reactivation manifests as acute myocarditis or cardiomyopathy. In patients who already have cardiac damage due to chronic infection, reactivation can lead to new, acute inflammation or worsening congestive heart failure.13,14 Signs and symptoms of systemic infection may also be present. Reactivation of cardiac disease may occur with or without neurological disease.
Histopathologically, microglial nodules and multifocal gliosis with nests of amastigotes can be seen in brain tissue (Fig. 53-1). Cardiac involvement may manifest as a mild inflammatory reaction with focal, intrafascicular or endomysial infiltrates, and amastigotes with localized or diffuse inflammation can also be demonstrated in the bridging fibrous fascicles from endocardial and epicardial tissue (Fig. 53-2).
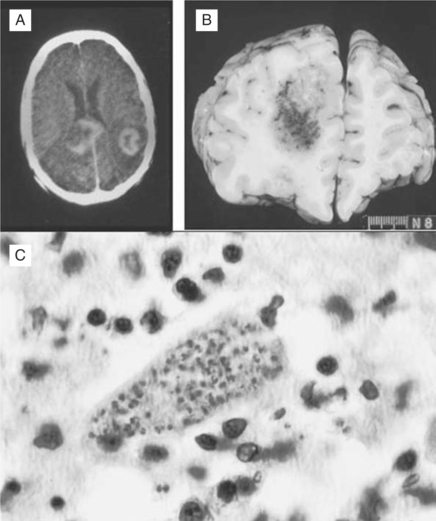
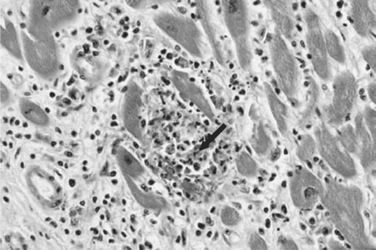
Diagnosis
Definitive diagnosis of T. cruzi requires demonstration of the organisms. Microscopy of blood smears is most useful during the acute stage and in symptomatic reactivation of chronic infection, when large numbers of parasites circulate in the bloodstream. In a phase contrast microscope, the motility of live trypomastigotes can easily be seen on a wet mount. In addition to Giemsa-stained blood smears, parasites may also be visualized in lymph nodes, bone marrow, pericardial fluid, intestinal mucosa, cerebrospinal fluid, and CNS mass lesions (Fig. 53-3). Certain microbiological tests, such as xenodiagnosis (recovering the parasite after inoculation of laboratory-raised insect vectors) or blood culture in liquid medium are more sensitive than direct visualization methods, but may take 2–8 weeks to become positive and are available only in highly specialized research settings.15 When reactivation is suspected, patients may require therapy while awaiting the results of definitive testing. Because of their increased sensitivity, these tests are most useful in the chronic stages of T. cruzi infection, when the level of parasitemia is low, and in detection of indeterminate infection. Polymerase chain reaction (PCR) of blood also has been shown to be an extremely sensitive method to detect trypanosomes in the blood but it is not yet available in most clinical facilities.16,17
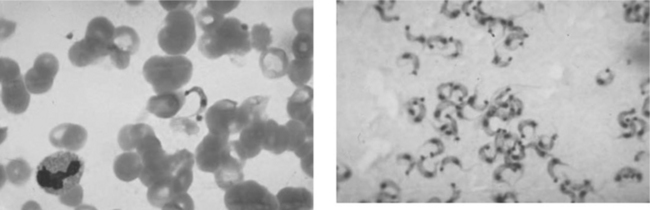
Serological tests to detect IgG antibody responses to T. cruzi are useful for diagnosis of chronically infected patients, to screen blood donors, and for seroepidemiological studies. IgM detection does not differentiate exposure from infection, and both IgM and IgG may be absent in HIV patients with trypanosomiasis.18,19 Multiple tests are available in endemic areas and several ELISA-based tests can be found in the United States. In the absence of therapy, the IgG remains positive for life, although the level may fall below the threshold of detection in advanced HIV disease. For this reason, the diagnosis of Chagas’ disease should not be discarded based on negative serological tests if the patient comes from an endemic region and has clinical findings compatible with Chagas’ disease. In such cases, direct parasitological testing (e.g., microscopic examination of blood or CSF for trypomastigotes) is the best diagnostic strategy.
Chagas’ disease shares a similar presentation to toxoplasmosis when it presents in the CNS, making this differentiation difficult. Chagomas have a predilection for subcortical white matter, with occasional gray matter involvement. The infection tends to cause extensive, diffuse hemorrhage.20 In contrast, cerebral toxoplasmosis is usually found in the cerebral cortex and basal ganglia.18,21 The differential diagnosis in endemic areas includes other entities such as lymphoma, bacterial abscesses, and tuberculosis. PCR plus microscopy of CSF or brain biopsy is often necessary to establish the etiology unequivocally.
Treatment
The two effective treatment regimens of Chagas’ disease is indicated during acute infection and in the case of reactivation. Although the role of therapy during the chronic or indeterminate phase is unclear, treatment is often recommended in cases of infection of less than 10 years’ duration and without overt cardiomyopathy, megacolon, or megaesophagus.22 Treatment under these conditions is administered in the hope of limiting progression to end-organ damage. The two effective treatment regimens are nifurtimox (8–10 mg kg−1 day−1 divided three times per day) for 90–120 days or benznidazole (5–7 mg kg−1 day−1 in two divided doses) for 30–90 days. Children 1–10 years of age require 15–20 mg kg−1 day−1 and 12–16 year olds should be given 12.5–15 mg kg−1 day−1, divided in four doses. The pediatric dose of benznidazole is 10 mg kg−1 day−1 in two doses. Common side effects of benzidazole include hypersensitivity, bone marrow depression, and peripheral neuropathy, and may require suspension of treatment with benznidazole. Weight loss, GI distress, and psychiatric disturbance may result from treatment with nifurtimox.23 No interaction between antitrypanosomal drugs and antiretroviral drugs has been reported. Availability of these drugs varies by country. Only nifurtimox, produced by Bayer in Germany, is available in the United States. This drug can be obtained from the Centers for Disease Control and Prevention (CDC) Drug Service. Benznidazole is produced in Brazil by Roche.
Therapy of reactivation disease is considered successful if there is resolution of parasitemia or parasitic tissue infection. With successful treatment, the IgG can become negative, but seroconversion may not be a reliable indicator of cure: in the presence of HIV co-infection antibody may decrease over time despite persistence of active infection. People living with HIV may be at risk for incomplete cure and recurrent reactivation disease. Although no studies have investigated secondary prophylaxis rigorously, it is standard practice in Brazil to administer secondary prophylaxis after reactivation disease with benznidazole at 5 mg kg−1 day−1 three times per week for life. Benznidazole is highly toxic and difficult to tolerate for long-term administration. Other alternatives include allopurinol or triazole derivatives, although their efficacy has not been documented.11,12 A proposed algorithm for the diagnosis and treatment of Chagas’ disease is shown in Figure 53-4.
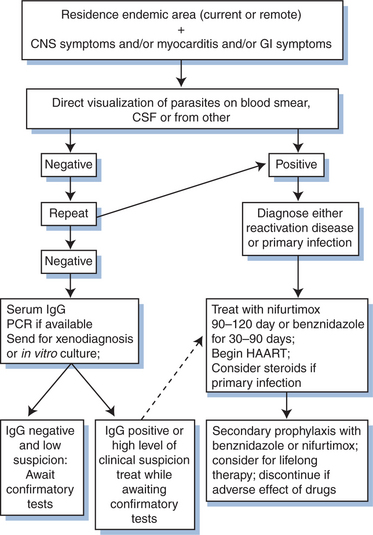
Primary prophylaxis for Chagas’ disease is not utilized: the efficacy of such prophylaxis is unknown, and there is no convenient and well-tolerated agent that is likely to be effective.18
Prognosis
Even with therapy, people with HIV co-infection and CNS reactivation rarely survive beyond 3 months. In small case series, outcomes appear to be improved with treatment early in the course of the disease.12 In Brazil, Chagas’ disease was added to the list of AIDS-defining illnesses in 2004.10 Antiretroviral therapy in addition to antitrypanosomal agents may improve the outcomes of chagasic meningoencephalitis and reactivation cardiac disease and should be utilized. There are no reports yet of related immune reconstitution syndromes.
LEISHMANIASIS
Leishmania species are obligate intracellular parasites that cause a broad spectrum of clinical disease in man that can generally be classified as visceral, mucosal, or cutaneous leishmaniasis. Visceral leishmaniasis (VL) is caused by L. infantum (in Europe, the Middle East, and Africa), L. donovani (in Asia) or L. chagasi (in South America) and is the clinical form that has the strongest interaction with HIV co-infection. It is spread by insect vectors and via blood, usually due to needle sharing among intravenous drug users.24,25 Asymptomatic infection is very common and the incubation period from initial disease to infection is 2–6 months or longer. A minority of immunocompetent patients with subclinical infection progress to develop symptomatic disease. In contrast, symptomatic disease is frequent when severely immunosuppressed patients become infected. The interaction between HIV and Leishmania has been best described in Europe where diagnostic resources are available. In the other regions where HIV and Leishmania coexist, notably Brazil and east Africa, less data are available.26
Clinical Presentation
With HIV-associated immunosuppression, exposed individuals progress to VL more frequently than immunocompetent hosts. HIV-infected patients may present with unusual manifestations. HIV co-infection increases the risk of VL 100- to 1000-fold in endemic areas of Europe, where the disease is found predominantly in HIV-co-infected patients with CD4+ T-lymphocyte cell counts less than 200 cells/mm3.27,28 The triad of fever, hepatomegaly, and splenomegaly is the typical manifestation found in HIV-infected patients with CD4+ T-lymphocyte cell counts >50 cells/mm3. Fever alone may be the presenting symptom of VL among patients with extremely low CD4+ T-lymphocyte cell counts. Anemia, thrombocytopenia, and leucopenia are common manifestations of symptomatic disease, even when the classic triad is absent.29,30 Through uncontrolled hematogenous dissemination, the pathogens are able to infect atypical locations, leading to unusual clinical manifestations. Lesions have been documented to involve the gastrointestinal and respiratory tracts, peritoneum, pleura, eyes, skin, and tonsils.29,31–33 Skin lesions of disseminated VL may mimic Kaposi sarcoma (Fig. 53-5).34,35 Strains that only cause cutaneous infection in normal hosts may visceralize when HIV co-infection is present.36
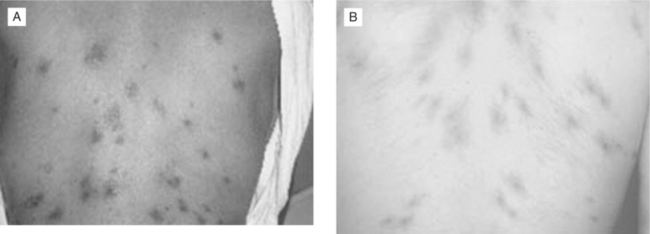
Although VL is not an AIDS-defining illness, symptomatic disease usually occurs in patients with very low CD4+ T-lymphocyte counts. The presentation of leishmaniasis often coincides with other opportunistic infections, and thus clinicians should consider the possibility that VL is not the only process responsible for a complex clinical syndrome.27 In studies from Europe, when the diagnosis of VL was made, 42–68% of patients had additional concurrent opportunistic infections.28
Diagnosis
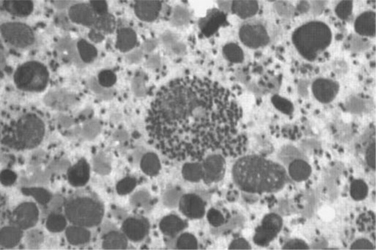
The gold standard for diagnosis of leishmaniasis is the demonstration of amastigotes on bone marrow, liver, or spleen biopsy (Fig. 53-6). Where possible, bone marrow aspirate is preferred over spleen aspirate for safety concerns. Specimens from individuals with low CD4+ T-lymphocyte cell counts are more frequently positive by this method compared to patients with higher CD4+ T-lymphocyte cell counts due to the higher parasite burden.29 With advanced HIV disease, hypocellular bone marrow may make the diagnosis more difficult since these organisms are found intracellularly. PCR tests of blood can also be useful although these tests are not standardized for sensitivity or specificity.37,38 PCR can also be used to speciate the organism.39 In individuals with low CD4+ T-lymphocyte cell counts, staining of the buffy coat of the blood or a blood culture with Leishmania-specific media (Schneiders or NNN media) may be used for diagnosis.40,41 In cases of disseminated disease with infection in atypical locations, the organisms may only be visible at the sites of disseminated disease.29
The three main serological assays utilized for diagnosing leishmaniasis include the indirect immunoflourescent antibody test (IFAT), ELISA to detect antibodies to the cloned antigen rK39, and Western blot.28 If the tissue diagnosis cannot be obtained, at least two different serological tests should be used in HIV-positive persons. Leishmanial antigens should be freshly prepared to increase sensitivity.27 A summary of diagnostic methods for leishmaniasis in the presence of HIV co-infection is found in Table 53-1.
Table 53-1 Diagnostic methods for cases of Leishmania/HIV co-infection
| Method | % Positive |
|---|---|
| Microscopic examination of bone marrow smear | 8627 |
| Microscopic examination of blood smear | 4527 |
| Microscopic examination of tissue smear (skin, GI tract, liver, spleen) | 8727 |
| Hemoculture using Schneider’s or Novy–McNeal–Nicolle (NNN) media | 72–8740,52 |
| IFAT (indirect immunoflourescence antibody test) | 11–5827,40,99,100 |
| ELISA (anti-rK39) | 22–6027,99,100 |
| Western blot | 7127 |
Treatment
In the HIV-co-infected population, drug-associated toxicities are severe and can be fatal. Where Leishmania retain susceptibility to antimonials, these drugs are as effective as amphotericin B. In HIV-co-infected patients in Europe, cure rates were ∼75% in both the meglumine arm and the amphotericin B arm in a study among subjects who were able to complete the full course of therapy. However, in a series of studies from Spain, 19/63 (30%) HIV-positive patients who were assigned to the meglumine arm were unable to complete therapy due to adverse side effects.42,43 In Africa, the adverse reactions may be even more severe. In Ethiopia, SSG was associated with a 90% cure rate, but a 6% death rate in the first month of therapy among HIV-infected participants. In the miltefosine arm of this study, poorer efficacy was observed, yet only 1% died in the first month of therapy.44 The higher risk of death is attributed to antimonial toxicity, although the precise causes of death were not identified. SSG is produced by GlaxoSmithKline and can be obtained in the United States through the CDC Drug Service. Meglumine is made by Aventis in France but is not available in the US.
The best-tolerated treatment option for HIV-positive individuals is the liposomal formulation of amphotericin B (3 mg kg−1 day−1).42 Several dosing schedules have been shown to be effective including administration for 10 days or for 5 days followed by doses on day 14 and 21. It has minimal toxicity and is usually as effective as or more effective than other options. In Europe, where leishmaniasis occurs almost exclusively as a complication of immunosuppression, liposomal amphotericin B or amphotericin B lipid complex is the standard therapy. Amphotericin B lipid complex is licensed in the United States as first-line therapy for VL.
In resource-limited settings where leishmaniasis is endemic, antileishmanial drugs cannot be widely used due to prohibitively high costs and the requirement for intra-venous administration. A new treatment option, miltefosine (2.5 mg kg−1 day−1 for 28 days) is an oral therapy for VL that is effective against organisms resistant to the antimony compounds. Miltefosine is effective in immunocompetent hosts,45 but efficacy in HIV-co-infected hosts has not been well-documented. A small study in Ethiopia showed miltefosine to be less effective, although better tolerated, than the antimonial compounds in HIV-positive individuals.44 In a review of the cases of compassionate use for HIV-infected individuals who had contraindications to amphotericin B or failed previous therapy, miltefosine was associated with a 41% cure rate. It was also well tolerated, with no dose-limiting side effects.46 Other isolated case reports have demonstrated miltefosine efficacy with HIV co-infection either alone or in combination with other agents.47,48 Miltefosine has significant reproductive toxicity. It cannot be used in pregnant women and women are recommended to use effective contraception during and for 2 months after therapy. It is produced by Zentaris in Germany and has been distributed in Europe on a compassionate use protocol, but is not currently available in the United States.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


