Inherited disorders of platelet function are characterized by highly variable mucocutaneous bleeding manifestations. The platelet dysfunction arises by diverse mechanisms, including abnormalities in platelet membrane glycoproteins, granules and their contents, platelet signaling and secretion mechanisms: thromboxane production pathways and in platelet procoagulant activities. Platelet aggregation and secretion studies using platelet-rich plasma currently form the primary basis for the diagnosis of an inherited platelet dysfunction. In most such patients, the molecular and genetic mechanisms are unknown. Management of these patients needs to be individualized; therapeutic options include platelet transfusions, 1-desamino-8 d -arginine vasopressin (DDAVP), recombinant factor VIIa, and antifibrinolytic agents.
Key points
- •
Inherited disorders of platelet function are characterized by highly variable mucocutaneous bleeding manifestations that are mild to moderate in most patients.
- •
The platelet dysfunction in these patients arises by diverse mechanisms, including abnormalities in platelet membrane glycoproteins, granules, signaling and secretion mechanisms, and procoagulant activities.
- •
In the vast majority of patients suspected to have an inherited platelet function defect, the molecular and genetic mechanisms are unknown.
- •
Platelet aggregation and secretion studies using platelet-rich plasma form the primary basis for the diagnosis of an inherited platelet dysfunction in most patients.
- •
The therapeutic options in these patients include platelet transfusions, DDAVP, recombinant factor VIIa, and antifibrinolytic agents.
Platelet function in hemostasis
Following injury to the blood vessel, platelets adhere to exposed subendothelium by a process (adhesion) that involves, among other events, the interaction of a plasma protein, von Willebrand factor (vWF), and a specific glycoprotein complex on the platelet surface, glycoprotein (GP) Ib-IX-V (GPIb-IX) ( Fig. 1 ). This interaction is particularly important for platelet adhesion under conditions of high shear stress. Adhesion is followed by recruitment of additional platelets that form clumps, a process called aggregation (cohesion). This platelet-platelet interaction involves binding of fibrinogen to specific platelet surface receptors, a complex composed of GPIIb-IIIa (integrin αIIbβ3). Resting platelets do not bind fibrinogen and platelet activation induces a conformational change in the GPIIb-IIIa complex that leads to fibrinogen binding. Activated platelets release the contents of their granules (secretion), including ADP and serotonin from the dense granules, which causes the recruitment of additional platelets. In addition, contents of the α-granules and other vesicles are also released. Moreover, platelets play a major role in coagulation mechanisms; several key enzymatic reactions occur on the platelet membrane lipoprotein surface. During platelet activation, the negatively charged phospholipids, especially phosphatidylserine, become exposed on the platelet surface, an essential step for accelerating specific coagulation reactions by promoting the binding of coagulation factors involved in thrombin generation (platelet procoagulant activity).
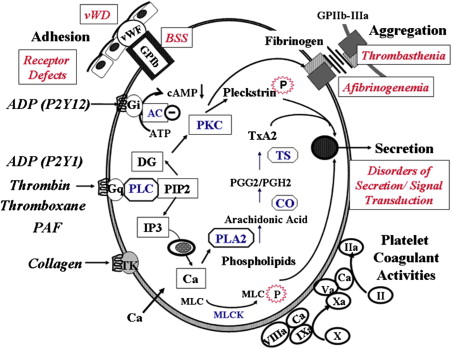
A number of physiologic agonists interact with specific receptors on the platelet surface to induce responses, including a change in platelet shape from discoid to spherical (shape change), aggregation, secretion, and thromboxane A 2 (TxA 2 ) production. Other agonists, such as prostacyclin, inhibit these responses by increasing cyclic AMP levels (cAMP). Binding of agonists to platelet receptors initiates the production or release of several intracellular messenger molecules, including products of hydrolysis of phosphoinositide (PI) by phospholipase C (diacylglycerol and inositol 1,4,5-triphosphate [InsP 3 ]), TxA 2 , and cyclic nucleotides (cAMP) (see Fig. 1 ). These induce or modulate the various platelet responses of Ca 2+ mobilization, protein phosphorylation, aggregation, secretion, and thromboxane production. The interaction between the platelet surface receptors and the key intracellular enzymes (eg, phospholipases A 2 and C, adenylyl cyclase) is mediated by a group of proteins that bind and are modulated by guanosine triphosphate (G proteins). As in most secretory cells, platelet activation results in an increase in cytoplasmic ionized calcium concentration; InsP 3 functions as a messenger to mobilize Ca 2+ from intracellular stores. Diacylglycerol activates protein kinase C (PKC), and this results in the phosphorylation of several proteins. PKC activation plays a major role in regulating responses, including platelet secretion and the activation of GPIIb-IIIa. Numerous other mechanisms, such as activation of tyrosine kinases and phosphatases, are also triggered by platelet activation. Inherited or acquired defects in these platelet mechanisms may lead to impairment of the platelet role in hemostasis.
Platelet function in hemostasis
Following injury to the blood vessel, platelets adhere to exposed subendothelium by a process (adhesion) that involves, among other events, the interaction of a plasma protein, von Willebrand factor (vWF), and a specific glycoprotein complex on the platelet surface, glycoprotein (GP) Ib-IX-V (GPIb-IX) ( Fig. 1 ). This interaction is particularly important for platelet adhesion under conditions of high shear stress. Adhesion is followed by recruitment of additional platelets that form clumps, a process called aggregation (cohesion). This platelet-platelet interaction involves binding of fibrinogen to specific platelet surface receptors, a complex composed of GPIIb-IIIa (integrin αIIbβ3). Resting platelets do not bind fibrinogen and platelet activation induces a conformational change in the GPIIb-IIIa complex that leads to fibrinogen binding. Activated platelets release the contents of their granules (secretion), including ADP and serotonin from the dense granules, which causes the recruitment of additional platelets. In addition, contents of the α-granules and other vesicles are also released. Moreover, platelets play a major role in coagulation mechanisms; several key enzymatic reactions occur on the platelet membrane lipoprotein surface. During platelet activation, the negatively charged phospholipids, especially phosphatidylserine, become exposed on the platelet surface, an essential step for accelerating specific coagulation reactions by promoting the binding of coagulation factors involved in thrombin generation (platelet procoagulant activity).
A number of physiologic agonists interact with specific receptors on the platelet surface to induce responses, including a change in platelet shape from discoid to spherical (shape change), aggregation, secretion, and thromboxane A 2 (TxA 2 ) production. Other agonists, such as prostacyclin, inhibit these responses by increasing cyclic AMP levels (cAMP). Binding of agonists to platelet receptors initiates the production or release of several intracellular messenger molecules, including products of hydrolysis of phosphoinositide (PI) by phospholipase C (diacylglycerol and inositol 1,4,5-triphosphate [InsP 3 ]), TxA 2 , and cyclic nucleotides (cAMP) (see Fig. 1 ). These induce or modulate the various platelet responses of Ca 2+ mobilization, protein phosphorylation, aggregation, secretion, and thromboxane production. The interaction between the platelet surface receptors and the key intracellular enzymes (eg, phospholipases A 2 and C, adenylyl cyclase) is mediated by a group of proteins that bind and are modulated by guanosine triphosphate (G proteins). As in most secretory cells, platelet activation results in an increase in cytoplasmic ionized calcium concentration; InsP 3 functions as a messenger to mobilize Ca 2+ from intracellular stores. Diacylglycerol activates protein kinase C (PKC), and this results in the phosphorylation of several proteins. PKC activation plays a major role in regulating responses, including platelet secretion and the activation of GPIIb-IIIa. Numerous other mechanisms, such as activation of tyrosine kinases and phosphatases, are also triggered by platelet activation. Inherited or acquired defects in these platelet mechanisms may lead to impairment of the platelet role in hemostasis.
Inherited disorders of platelet function: an overview
Disorders of platelet function are characterized by highly variable mucocutaneous bleeding manifestations and excessive hemorrhage following surgical procedures or trauma. Spontaneous hemarthrosis and deep hematomas are distinctly unusual in patients with platelet defects. In general, most patients have mild to moderate bleeding manifestations. Most patients, but not all, have a prolonged bleeding time. Platelet aggregation and secretion studies using platelet-rich plasma (PRP) provide evidence for platelet dysfunction but are, in general, neither predictive of severity of clinical manifestations nor the molecular mechanisms. Defects in platelet function may be inherited or acquired, with the latter being far more commonly encountered. The platelet dysfunction in these patients arises by diverse mechanisms. In the vast majority of patients suspected to have an inherited platelet function defect based on family studies, the molecular and genetic mechanisms are unknown.
Box 1 provides a classification of inherited disorders associated with impaired platelet function, based on the platelet function or responses that are abnormal (see Fig. 1 ). Of note, not all of them are caused by a defect in the platelets per se. Some, such as von Willebrand disease (vWD) and afibrinogenemia, result from deficiencies of plasma proteins essential for platelet adhesion or aggregation. Some of these disorders are distinctly rare, but they shed enormous light on platelet physiology. Moreover, in many patients with inherited abnormal platelet aggregation responses, the underlying molecular mechanisms remain unknown. In patients with defects in platelet–vessel wall interactions (adhesion disorders), adhesion of platelets to subendothelium is abnormal. The 2 disorders in this group are vWD, in which there is a deficiency or abnormality in plasma vWF, and the Bernard-Soulier syndrome (BSS), in which platelets are deficient in GPIb (and GPV and GPIX); in both disorders, platelet-vWF interaction is compromised. Binding of fibrinogen to the GPIIb-IIIa complex is a prerequisite for platelet aggregation. Disorders characterized by abnormal platelet-platelet interactions (aggregation disorders) arise because of a severe deficiency of plasma fibrinogen (congenital afibrinogenemia) or because of a quantitative or qualitative abnormality of the platelet membrane GPIIb-IIIa complex, which binds fibrinogen (Glanzmann thrombasthenia). Patients with defects in platelet secretion and signal transduction are a heterogeneous group lumped together for convenience of classification rather than based on an understanding of the specific underlying abnormality. The major common characteristics in these patients, as currently perceived, are abnormal aggregation responses and an inability to release intracellular granule (dense) contents on activation of PRP with agonists, such as ADP, epinephrine, and collagen. In general, in platelet studies, the primary wave of aggregation is present but the second wave is blunted or absent. In these patients, the platelet dysfunction arises from diverse mechanisms. A small proportion have a deficiency of dense granule or their stores (storage pool deficiency). In other patients, the impaired secretion results from aberrations in the signal transduction events or other mechanisms, such as in pathways leading to thromboxane synthesis, in mechanisms that govern end-responses of secretion and aggregation. The findings on the aggregation studies are nonspecific and it is difficult to conclude a specific abnormality from the tracings. Another group consists of patients who have an abnormality in interactions of platelets with proteins of the coagulation system; the best described is the Scott syndrome, which is characterized by impaired transmembrane migration of procoagulant–phosphatidylserine during platelet activation. Defects related to platelet cytoskeletal or structural proteins may also be associated with platelet dysfunction. Recent studies document impaired platelet function associated with mutations in transcription factors (eg, RUNX1, GATA1, FLI-1) that regulate expression of important platelet proteins. In addition to these groups, there are patients who have abnormal platelet function associated with systemic disorders, such as Down syndrome and the May-Hegglin anomaly, in which the specific aberrant platelet mechanisms are unclear. The prevalence and relative frequencies of the various platelet abnormalities described previously remain unknown. Disorders related to platelet membrane glycoproteins are described in the article by Drs Reyhan Diz-Kücükkaya and José A. López, elsewhere in this issue. Other inherited platelet disorders are reviewed here.
- 1.
Defects in platelet–vessel wall interaction (disorders of adhesion)
- a.
von Willebrand disease (deficiency or defect in plasma vWF)
- b.
Bernard-Soulier syndrome (deficiency or defect in GPIb)
- a.
- 2.
Defects in platelet-platelet interaction (disorders of aggregation)
- a.
Congenital afibrinogenemia (deficiency of plasma fibrinogen)
- b.
Glanzmann thrombasthenia (deficiency or defect in GPIIb-IIIa)
- a.
- 3.
Disorders of platelet secretion and abnormalities of granules
- a.
Storage pool deficiency (δ, α, αδ)
- b.
Quebec platelet disorder
- a.
- 4.
Disorders of platelet secretion and signal transduction
- a.
Defects in platelet-agonist interaction (receptor defects) (ADP, thromboxane A 2 , collagen, epinephrine)
- b.
Defects in G-proteins (Gαq, Gαs, Gαi abnormalities)
- c.
Defects in phosphatidylinositol metabolism and protein phosphorylation
Phospholipase C-β2 deficiency
PKC- deficiency
- d.
Abnormalities in arachidonic acid pathways and thromboxane A 2 synthesis
Phospholipase A2 deficiency
Cyclooxygenase deficiency
Thromboxane synthase deficiency
- a.
- 5.
Disorders of platelet coagulant-protein interaction (Scott syndrome)
- 6.
Defects related to cytoskeletal/structural proteins
- a.
Wiskott-Aldrich syndrome
- b.
β1 tubulin deficiency
- c.
Kindlin-3 deficiency (leukocyte adhesion defect-III)
- a.
- 7.
Abnormalities of transcription factors leading to functional defects
- a.
RUNX1 (Familial platelet dysfunction with predisposition to acute myelogenous leukemia);
- b.
GATA-1
- a.
Disorders of platelet secretion, granules, and signal transduction
Patients lumped in this remarkably heterogeneous group of platelet secretion defects generally manifest decreased aggregation and absence of the second wave of aggregation on stimulation of PRP with ADP and epinephrine, and impaired secretion of dense granule contents; responses to collagen, thromboxane analog (U46619), arachidonic acid, and thrombin receptor peptides may also be impaired. Simplistically in these patients, platelet secretion and function are abnormal either because the granules or their contents are diminished (SPD) or when there are aberrations in the signaling or activation mechanisms that lead to aggregation and secretion on platelet activation (see Fig. 1 ).
During the late 1960s, several investigators described patients with bleeding disorders associated with abnormalities in platelet aggregation induced by collagen, ADP, and epinephrine. In some of these patients, the abnormal platelet responses were attributed to a defect in the secretion of ADP. In 1969, Weiss and colleagues reported a family with impaired platelet aggregation whose platelets had decreased amounts of ADP. Holmsen and Weiss subsequently established that the defect in this family was a deficiency in the nonmetabolic pool of ADP that is stored in the dense granules, leading to the entity being called “storage pool deficiency” (SPD). In 1982, Rao and colleagues reported 5 patients with a lifelong bleeding diathesis whose platelets had decreased aggregation, and diminished secretion of dense granule and acid hydrolase secretion even though their platelets had normal granule stores and thromboxane production. Such patients without SPD or defective TxA 2 production were subsequently referred to as “primary secretion defects” and described in several reports. Such patients are more common than those with thrombasthenia, BSS, SPD, or defects in TxA 2 production. In some of them, there is evidence for abnormalities in the specific signaling proteins or events that precede aggregation and secretion.
Disorders of platelet granules
SPD
The term SPD now encompasses patients with deficiencies in the platelet contents of dense granules (δ-SPD), α-granules (α-SPD) (gray platelet syndrome), or both types of granules (αδ-SPD). Another α-granule disorder is the Quebec platelet disorder (QPD), which is associated with abnormal proteolysis of several α-granule proteins.
δ-SPD
Patients with δ-SPD have a mild to moderate bleeding diathesis associated with a prolonged bleeding time. In the platelet studies, the second wave of aggregation in response to ADP and epinephrine is absent or blunted, and the collagen response is markedly decreased ( Fig. 2 ). Both impaired and normal aggregation responses to arachidonic acid have been noted. These conflicting observations may be related to the severity of platelet ADP deficiency. The responses to epinephrine may also be variable; a second wave of aggregation has been noted in some patients. Interestingly, δ-SPD has been documented in some patients with prolonged bleeding times and normal aggregation responses. Normal bleeding times have also been observed in δ-SPD. Thrombin-induced secretion of acid hydrolases is impaired in SPD platelets; this is corrected by addition of exogenous ADP, suggesting that it is secondary to the ADP deficiency and secretion.
Normal platelets possess 3 to 8 dense granules (each 200–300 nm in diameter). Under the electron microscope, platelet-dense granules are decreased in δ-SPD. Other methods to demonstrate a decrease in the dense granules include fluorescence microscopy after staining platelets with mepacrine (quinacrine) and specific staining by uranyl ions (uranaffin reaction). By direct biochemical measurements, the total platelet and granule ATP and ADP contents are decreased along with other dense granule constituents, calcium, pyrophosphate, and serotonin. Two-thirds of platelet ATP and ADP resides in the dense granules with a smaller amount in the metabolic pool; dense granules have proportionally greater ADP than ATP. Thus, in δ-SPD platelets, the ratio of total ATP to ADP increases (>2.5) compared with normal platelets.
Incubation of normal platelets with 14 C-serotonin results in its incorporation into dense granules. This serotonin is subsequently secreted on activation, providing a method to assess dense granule secretion. In normal platelets, the incorporated 14 C-serotonin remains in the platelet over 4 to 6 hours of incubation, being protected from the mitochondrial monoamine oxidases because of its sequestration in the dense granules. In SPD platelets, the uptake of 14 C serotonin is normal; however, this serotonin is metabolized by the cytoplasmic monoamine oxidases to 5-hydroxyindoleacetic acid and 5-hydroxytryptophol, resulting in the loss of the radioactive label from the platelets.
Other Abnormalities in δ-SPD
Other associated abnormalities have been described in some patients, including in synthesis of prostaglandins, thromboxane A2 and malondialdehyde, and in platelet procoagulant activity (prothrombinase activity) in association with an inability of δ-SPD platelets to maintain elevated intracellular Ca 2+ levels. Both the Ca 2+ defect and the decreased prothrombinase activity are corrected by addition of exogenous ADP, indicating that dense granule constituents may play a role in these responses.
δ-SPD has been reported in association with other inherited disorders, such as the Hermansky-Pudlak syndrome (HPS) (a combination of oculocutaneous albinism, nystagmus, and increased reticuloendothelial ceroid), the Chediak-Higashi syndrome (CHS), the Wiskott-Aldrich syndrome (WAS), the thrombocytopenia-absent-radii (TAR) syndrome, and the Griscelli syndrome. The simultaneous occurrence of δ-SPD and defects in skin pigment granules, as in the HPS, point to the interrelatedness of the 2 kinds of granules (dense granules and melanosomes) with respect to genetic control. This concept has been strongly supported by studies in animal models that combine platelet defects with pigment disorders.
Pathogenesis
Studies in animal models suggest that dense granule abnormalities may occur by different mechanisms and involve defects at the megakaryocyte level. Dense granules are absent in megakaryocytes of CHS cattle that also have a pigmentary disorder, suggesting defective organelle development. In some mouse models (also having a pigment disorder), platelets have been found to have a substantial or normal number of mepacrine-positive granules, suggesting that there may be functional defect leading to impaired localization of the nucleotides to the granules, rather than a lack of granule formation. In line with this, some patients with HPS and δ-SPD have markedly decreased dense granules, suggesting an abnormality in granule development, whereas some patients without HPS have had the presence of uranaffin and mepacrine-positive granules but with a lack of dense core (“empty granules”), suggesting a more qualitative granule defect. Dense granule membranes possess the lysosomal proteins LAMP2 (lysosomal-associated membrane protein-2) and CD63 (granulophysin or LAMP3), as well as P-selectin and GPIIb-IIIa. Granulophysin has been shown to be deficient in some HPS platelets. Studies with antigranulophysin antibody demonstrated the presence of a normal number of platelet granules in 2 patients with SPD who were not albino ; these patients, thus, have the granules but with reduced contents.
A substantial amount of our information in SPD has been obtained from patients with HPS, which is characterized by oculocutaneous albinism, platelet SPD, and lipofuscinosis. There is a large group of patients with HPS in northwest Puerto Rico where HPS occurs in 1 of every 1800 individuals (gene frequency 1 in 21). There are at least 9 known HPS-causing genes leading to 9 subtypes of human HPS, with most of the patients being in HPS-1 and from Puerto Rico. There are more than 15 mouse models of HPS reported to date; many of these constitute models for the human subtypes. Together, the human and mouse models have been an invaluable source of basic information of vesicle formation and trafficking. The human HPS subtypes are autosomal recessive and the heterozygotes have no clinical findings. In addition to the albinism that is variable among the HPS subtypes, most patients have congenital nystagmus and decreased visual acuities. Moreover, approximately 15% of the patients develop granulomatous colitis, which resembles Crohn disease in pathology and in response to treatment. Another manifestation is pulmonary fibrosis, a crippling end result of an inflammatory process. An antifibrotic agent pirfenidone appears to slow the progression of pulmonary fibrosis. Of the multiple HPS subtypes, HPS-1 is the most severe and prevalent form of HPS. It arises from mutations in the HPS-1 gene; the most frequent HPS-1 mutation is a 16-bp duplication in exon 15, although other mutations have been noted. In general, with one exception (HPS-2) caused by mutations in AP3B1, all of the HPS-causing genes encode novel proteins. The gene defective in HPS-2, AP3B1, codes for the β3A subunit of AP3, a heterotetrameric complex responsible for vesicle formation from the trans-Golgi network.
CHS is a rare autosomal recessive disorder characterized by SPD, oculocutaneous albinism, immune deficiency, neurologic dysfunction, and the presence of giant cytoplasmic inclusions in different cells. Patients with CHS have defective cytotoxic T and NK cell function. CHS arises from mutations in the lysosomal trafficking regulator ( LYST ) gene on chromosome 1. The protein coded by LYST interacts with several proteins, including the SNARE complex protein HRS and signaling proteins, and participates in intracellular membrane fusion reactions and vesicle trafficking.
α-granule SPD (Gray Platelet Syndrome)
The rubric “gray-platelet syndrome” (GPS) has been derived from the initial observation by Raccuglia in 1971 of a gray appearance of platelets with paucity of granules in peripheral blood smears from a patient with a lifelong bleeding disorder. Patients with GPS have an isolated deficiency of α-granule contents. They have a lifelong bleeding diathesis, mostly of autosomal recessive inheritance, mild thrombocytopenia, and prolonged bleeding time. Under the electron microscope, platelets and megakaryocytes reveal absent or markedly decreased α-granules. The platelets are deficient in α-granule proteins: platelet factor-4, β-thromboglobulin, vWF, thrombospondin, fibronectin, factor V, high molecular weight kininogen, and platelet-derived growth factor. Platelet aggregation responses have been variable. Responses to ADP and epinephrine were normal in most patients; in some patients, aggregation responses to thrombin, collagen, and ADP have been impaired. There is increased reticulin in the bone marrow from patients with GPS; this has been attributed to elevated plasma PDGF levels.
GPS is a markedly heterogeneous disorder characterized by thrombocytopenia, large platelet size, and deficiency of α-granules and their contents, which includes proteins synthesized by MK (eg, PF4, β-thromboglobulin) and those incorporated by endocytosis into the granules (eg, albumin, fibrinogen, immunoglobulin [Ig] G). The molecular mechanisms leading to α-granule deficiency in GPS and in αδ-SPD are unclear; they have been attributed to multiple mechanisms including failure of α-granule maturation during MK differentiation; of transport or targeting of proteins to α-granules; and synthesis of granule membranes. Proteomic studies in a patients with GPS suggested a failure to incorporate endogenously synthesized MK proteins into α-granules. Some patients with GPS have elevated plasma PF4, suggesting that PF4 synthesis was normal and the primary defect was impaired granule biogenesis with leakage of PF4. Several patients with decreased α-granule contents have had a mutation in transcription factor RUNX1, and PF4 is a transcriptional target of RUNX1. This suggests that in some patients, decreased platelet PF4 level may be due to a defect in transcriptional regulation and synthesis of PF4. GPS has been reported in a patient with an X-linked thrombocytopenia, thalassemia, and Arg216Gln mutation in GATA-1, a major regulator of megakaryopoiesis. GATA-1 knockdown mice have granule abnormalities with a decrease in PF4 mRNA in primary MK. Moreover, a mutation in VPS33B protein (a member of the Sec1/Munc18 protein family) involved in vesicle trafficking has been associated with human α-granule deficiency in the arthrogryposis multiplex congenital, renal dysfunction, and cholestasis (ARC) syndrome. More recently, a mutation in VPS16B gene has also been linked to α-granule deficiency in the ARC syndrome, and it appears that VPS16B and VPS33B interact with each other. Three groups have recently reported mutations in NBEAL2 , a gene that encodes for a protein linked to vesicle transport in neuronal cells. Together, these studies elegantly implicate defective vesicle transport as a major mechanism in GPS. Overall, it appears that the mechanisms are likely different in individual patients with GPS; the inheritance of α-granule deficiency has been autosomal recessive in most reports, and autosomal dominant or sex-linked in some.
Quebec Platelet Disorder
QPD is an autosomal dominant disorder associated with delayed bleeding and abnormal proteolysis of α-granule proteins due to increased amounts of platelet urokinase–type plasminogen activator arising from random duplication of PLAU gene. These patients have had normal to reduced platelet counts, proteolytic degradation of α-granule proteins, deficiency of α-granule multimerin (a factor V binding protein), and defective aggregation selectively with epinephrine. Platelet factor V, but not plasma factor V, is degraded along with several other α-granule proteins (fibrinogen, vWF, thrombospondin, osteonectin, fibronectin, and P-selectin). The platelets have increased fibrinolytic activity. They appear morphologically normal under the light microscope. Patients with QPD suffer from mucocutaneous bleeding, which is often delayed by 12 to 24 hours following injury and unresponsive to platelet transfusions but responsive to fibrinolytic inhibitors.
Disorders of secretion caused by defects in platelet signaling mechanisms
Signal transduction events encompass processes that are triggered by the interaction of agonists with specific platelet receptors and result in the activation of effectors, such as phospholipase C (PLC) and phospholipase A 2 (PLA2) (see Fig. 1 ), leading ultimately to end responses of aggregation and secretion. The link between the surface receptors for several agonists (eg, ADP, epinephrine, thrombin, and thromboxane A 2 ) and the effector enzymes is provided by G-proteins. Simplistically, if the key components of platelet signal transduction mechanisms are the surface receptors, the G-proteins, and the effectors, evidence now exists for specific platelet abnormalities at each of these levels.
Defects in Platelet-Agonist Interaction: Receptor Defects
Impaired platelet responses because of an abnormality in platelet surface receptors have been documented for TxA 2 , collagen, ADP, epinephrine, and PAF. Because ADP and TxA 2 play a synergistic role in amplifying the platelet responses to other agonists, patients with defects in the ADP or TxA 2 receptor have impaired responses to other agonists as well, including collagen and thrombin.
Thromboxane A 2 receptor defect
Mutations in the platelet TxA 2 receptor have been reported by Hirata and colleagues, who described an Arg 60 to Leu mutation in the first cytoplasmic loop of the TxA 2 receptor in 2 unrelated patients. This Arg 60 corresponds to a highly conserved basic residue among G-protein coupled receptors. These patients had a mild bleeding disorder with an autosomal dominant pattern of inheritance. Aggregation responses to several agonists were impaired with the exception of thrombin. The binding of TxA 2 analogs to platelets was normal. GTPase activity on activation with a TxA 2 analog, but not thrombin, was diminished, suggesting a defect in TxA 2 receptor–G-protein coupling. TxA 2 -induced activation of PLC (measured as Ca 2+ mobilization, and inositol trisphosphate and phosphatidic acid formation) was impaired, whereas PLA 2 activation and TxA 2 production were normal. Fewer than half the number of TxA 2 receptors are sufficient for irreversible aggregation with TxA 2 agonist. The finding that the aggregation responses were impaired in the heterozygous family members suggest a dominant negative effect of the mutation. Last, impaired aggregation response to TxA 2 has also been observed in patients without evidence for a defect of TxA 2 receptor.
Defects in platelet ADP receptors (P2Y1, P2Y2, and P2X1)
At least 3 receptors (P2Y1, P2Y12, P2X1) mediate ADP interaction with platelets (see the review by Brass and colleagues elsewhere in this issue). P2Y 1 receptors induce PLC activation, intracellular Ca 2+ mobilization, and shape change, whereas P2Y 12 receptors mediate inhibition of cAMP formation by adenylyl cyclase. ADP-induced platelet aggregation requires activation of both P2Y 1 and P2Y 12 receptors. P2X 1 receptors function as an ATP-gated and ADP-gated cation channel. Several patients have been described with P2Y 12 receptor abnormalities, characterized by blunted ADP-induced platelet aggregation responses, impaired ADP suppression of PGE 1 -induced elevation in cAMP, and normal ADP-stimulated shape change. Patients’ symptoms have ranged in severity, with moderately severe hemorrhage in association with surgery and trauma and prolonged bleeding times. Because released ADP potentiates the responses to other agonists, such as collagen and thromboxane A 2 , platelet aggregation in response to these agonists are also abnormal. Platelet binding of ADP or the ADP analog 2-methylthio-ADP was decreased in almost all of these patients.
The genetic defects have been defined in some of these patients. Three patients have had homozygous deletions in the P2Y 12 gene, resulting in premature termination and a lack of P2Y 12 protein. A homozygous missense mutation in the translation initiation codon was described in another patient, and another patient was reported to have a 2-nucleotide deletion (at amino acid 240) in one P2Y 12 gene allele, resulting in a frame shift and a premature stop codon. Although this last patient had one P2Y 12 allele with a normal coding region, the patient’s platelets lacked P2Y 12 receptors, suggesting repression of the normal allele or an unrelated abnormality in its transcriptional regulation. In contrast, platelets from the patient’s daughter had an intermediate number of ADP-binding sites, a normal platelet response to ADP, and one frame-shifted allele and one normal allele, suggesting that the mutant allele does not act in a dominant negative manner. Studies in yet another patient with abnormal ADP-induced aggregation revealed a compound heterozygous state encompassing an R256N substitution and an R265W substitution in the 2 alleles. The former mutation was in the sixth transmembrane domain and the latter was in the third extracellular loop of the receptor. Interestingly, platelet binding of 33 P-2MeS ADP was normal. In expression studies in Chinese hamster ovary cells, neither mutation affected the translocation of the P2Y 12 receptor to the cell surface, but ADP-induced inhibition of adenylyl cyclase was partially reduced, indicating a functional abnormality. In another study involving screening 92 patients with type 1 vWD, a heterozygous mutation in the second extracellular loop of P2Y 12 was identified in one patient and several family members ; this was associated with decreased 2MeS-ADP binding and modest defects in ADP-induced platelet aggregation. Thus, in some patients with vWD, the added platelet defect may contribute to the overall bleeding. Another heterozygous mutation, P258T in the third extracellular loop, has been described in association with a bleeding diathesis.
A defect in the P2X 1 purinergic receptor has been described in a 6-year-old patient with a history of petechiae, ecchymoses, and severe expistaxis. The patient had isolated impairment of ADP-induced platelet aggregation and was heterozygous for a deletion of a single leucine in a stretch of 4 leucine residues (351–354) in the second transmembrane domain of P2X 1 . The mutant protein apparently caused a dominant negative effect on P2X 1 -mediated calcium channel activity.
A preliminary report of one patient with a defect in the P2Y1 platelet receptor, which is coupled to PLC and ADP-induced calcium mobilization, described impaired platelet aggregation in response to ADP and other agonists.
Defects in platelet collagen receptors
Platelet-collagen interaction is important in hemostasis and thrombosis. Collagen is a substrate for platelet adhesion, a binding site for vWF in the extracellular matrix, and an agonist for platelet aggregation and secretion. Two receptors, GPVI and the integrin α2β1, mediate the platelet-collagen interaction.
GPVI, a member of the immunoglobulin gene superfamily, is associated with the Fc receptor γ chain in the platelet plasma membrane. Murine platelets lacking either the γ chain or Syk are unable to respond to collagen and the interaction of GPVI-deficient human platelets with either immobilized and soluble collagen is markedly impaired.
There are several reports of patients with congenital GPVI deficiency. They have had mild to severe mucocutaneous bleeding and their platelets failed to aggregate following stimulation with collagen despite the presence of normal amounts of α2β1, but retained some ability to adhere to collagen-coated surfaces. GPVI deficiency has also been reported in a patient with GPS. Several patients have been described whose plasma contained autoantibodies against GPVI that inhibited collagen-induced platelet function ; platelets of these patients have had little or no detectable GPVI on the platelet surface, and no mutations in GPVI were noted. In some patients, GPVI was restored on platelets following treatment of the immunologic disease. These studies suggest that the autoantibodies selectively deplete platelet GPVI.
The integrin α2β1 (GPIa-IIa, VLA-2) also serves as a platelet collagen receptor. Current models suggest that the initial platelet tethering of platelets is mediated by platelet GPIb-IX-V binding to vWF bound to collagen and this enables GPVI to bind to collagen and generate “outside-in” signals. These signals increase the affinity of α2β1 for collagen, resulting in a firm platelet adhesion to collagen. Several patients have been reported whose platelets lacked α2β1 and impaired interaction with collagen. Of 2 patients with bleeding diatheses, platelets from one contained approximately 15% to 25% of the normal amount of α2, failed to aggregate in response to collagen, and failed to adhere to collagen under both static and flow conditions, whereas platelets from the second patient lacked α2, as well as the α-granule protein thrombospondin, failed to aggregate in response to low concentrations of collagen, and adhered to, but did not spread on, collagen-coated surfaces. In 2 families with autosomal dominant thrombocytopenia and mild cutaneous bleeding, platelets had 38% to 63% of the normal amount of α2β1 and adhered poorly to collagen-coated surfaces, whereas collagen-induced platelet aggregation was either normal or only slightly decreased.
Defects in responsiveness to epinephrine
Epinephrine-induced aggregation, mediated by α2-adrenergic receptors (α 2 AR), may be decreased in some presumably healthy individuals. In one study, the second wave of aggregation was absent in 10% to 15% of healthy subjects. Studies in twins suggest that platelet α 2 AR expression is under genetic control. One family has been described in which several members had impaired aggregation and secretion in response to only epinephrine associated with decreased number of platelet α 2 AR. Three family members had a history of easy bruising with minimally prolonged bleeding times. Despite the diminished aggregation response, epinephrine inhibition of adenylate cyclase was normal, indicating that the receptor requirements for these 2 platelet responses are different. Although other families with an epinephrine defect have been reported, the relationship of the selective epinephrine defect to bleeding manifestations needs to be defined. The isolated impaired aggregation response to epinephrine has been reported in Quebec platelet disorder.
Defects in GTP-binding Proteins
GTP-binding proteins (consisting of α, β, and γ subunits) link surface receptors and intracellular enzymes and constitute a potential locus for aberrations leading to platelet dysfunction. Abnormalities involving Gαq, Gαi 2 , and Gαs have been described in human platelets.
Gαq deficiency
Gabbeta and colleagues described, in a patient with a mild bleeding disorder, abnormal aggregation and secretion in response to several agonists, and diminished GTPase activity (a reflection of α-subunit function) on platelet activation. The binding of 35 S-GTPγS to platelet membranes was diminished, and there was a selective decrease in platelet membrane Gαq with normal levels of Gα i2 , Gα 12 , Gα 13 , and Gα z . This patient had abnormalities in other downstream events, including activation of the GPIIb-IIIa complex, Ca 2+ mobilization, and release of arachidonic acid from phospholipids on platelet activation. Gαq coding sequence in this patient was normal and the Gαq mRNA levels are decreased in platelets but not neutrophils, which have normal responses and Gαq protein.
Gαs hyperfunction and genetic variation in extra-large Gαs
Activation of Gαs results in increased platelet cAMP levels and inhibition of platelet responses to activation. Two unrelated families have been described with inducible hyperactivity of Gαs. These patients had a bleeding diathesis, prolonged bleeding times, variable mental retardation, and mild skeletal malformations. Platelet aggregation responses to usual agonists were reportedly normal, but the platelets showed increased sensitivity to inhibition by agents that elevate cAMP levels. Exposure to PGE1, PGI2, or adenosine resulted in an enhanced increase in cAMP associated with an increased platelet Gαs protein. A heterozygous 36–base pair (bp) insertion and 2 bp substitutions were identified in the exon 1 of the extra-large Gαs (XLas) gene in these patients. Because XLas is not activated by activation of the usual platelet Gαs-coupled receptors, the mechanisms leading to the increased cAMP levels and the enhanced expression of Gαs protein in the patients remain unclear. Interestingly, 2.2% of control subjects also revealed the mutations detected in the patients, along with evidence of inducible Gαs hyperfunction and increased platelet Gαs protein.
Platelet Gαs deficiency has been described in a patient with pseudohypoparathyroidism Ib (PHPIb) in association with disturbed imprinting and altered methylation in the GNAS1 gene. The platelets showed decreased cAMP formation on Gαs receptor activation and Gαs protein deficiency; the Gαs coding sequence was normal. The investigators did not indicate whether the patient had a bleeding diathesis.
Gαi1 deficiency
The patient described with platelet Gαi1 deficiency presented with a bleeding disorder, and abnormal aggregation and dense granule secretion on activation with ADP, U46619, collagen, and epinephrine. Additionally, ADP-induced GPIIb-IIIa activation was diminished, indicating a role of Gαi1 in this response. A major platelet response mediated by Gαi is inhibition of adenylyl cyclase and cAMP levels. In the patient’s platelets, inhibition of forskolin-stimulated cAMP levels was absent on exposure to ADP, thrombin, or epinephrine. In contrast, Gαq-mediated responses (activation of PLC-β2, Ca 2+ mobilization, and pleckstrin phosphorylation) were normal. Platelet expression of Gαi1 was decreased by 75%; other members of the Gαi family (Gαi2, Gαi3, Gαiz) and Gαq were normal.
PLC-β2 Deficiency and Defects in Phospholipase C Activation, Calcium Mobilization and Protein Phosphorylation
Activation of PLC is an early event on platelet stimulation and leads to the formation of intracellular mediators InsP 3 and diacylglycerol (see Fig. 1 ) and to mechanisms such as Ca 2+ mobilization and PKC-induced protein phosphorylation. Defects in some of these specific responses have been documented. PKC-induced pleckstrin phosphorylation and cytoplasmic Ca 2+ mobilization play a major role in secretion and aggregation on activation. In one study of 8 patients with abnormal aggregation and secretion in response to several different agonists, receptor-mediated Ca 2+ mobilization and/or pleckstrin phosphorylation were abnormal in 7 of the 8 patients. These studies suggest that the later events of exocytosis or secretion per se are intact in these patients and impaired secretion and aggregation result from upstream abnormalities in early signaling events. True to this supposition, specific human defects at the level of PLC-β2, Gαq, and PKC- have been documented in these patients.
In another study, 8 patients have been described with decreased initial rates and extents of aggregation in response to weak agonists, ADP, epinephrine, and U44069; the investigators postulated defects in early platelet activation events to explain the abnormal responses. They subsequently demonstrated a defect in phosphatidylinositol hydrolysis and phosphatidic acid formation, and in pleckstrin phosphorylation in one patient.
Cytoplasmic Ca 2+ mobilization is an early response to platelet stimulation. Attention was, therefore, focused initially on this process to explain the impaired platelet function. Studies in 2 related patients with impaired aggregation and secretion responses revealed decreased peak Ca 2+ concentrations following activation with ADP, collagen, PAF, or thrombin, with abnormalities in intracellular release and in the influx of extracellular Ca 2+ . Formation of InsP3, the key intracellular mediator of Ca 2+ release, as well as diacylglycerol formation and pleckstrin phosphorylation, were diminished on platelet activation, indicating a defect in PLC activation (see Fig. 1 ). Human platelets contain at least 7 PLC isozymes in the quantitative order PLC-γ2 > PLC-β2 > PLC-β3 > PLC-β1 > PLC-γ1 > PLC-δ1 > PLC-β4. Studies in one of the patients with impaired PLC activation revealed a selective decrease in only the PLC-β2 isozyme. The decreased platelet PLC-β2 protein levels were associated with a normal coding sequence but with diminished PLC-β2 mRNA levels in platelets but not neutrophils, suggesting a hematopoietic lineage-specific defect in PLC-β2 gene regulation. These studies provide direct validation in human platelets of the importance in hemostasis of PLC-β2.
Abnormalities in these signal transduction pathways, including in phosphatidylinositol metabolism and protein phosphorylation, have been described in other patients, although the primary protein abnormality remains unknown. In one patient, the abnormal platelet aggregation was associated with decreased TxA 2 -induced InsP 3 formation but with normal GTPase activity and normal platelet TxA 2 receptors (including their cDNA sequence), suggesting that the abnormality in PLC activity was downstream of the surface receptor. Overall, these patients provide evidence for aberrations in the signal transduction pathways in patients with diminished platelet aggregation and secretion.
Defects in Protein Phosphorylation: PKC- Deficiency
PKC isozymes are a family of serine and threonine specific protein kinases that phosphorylate numerous proteins involved in signal transduction. PKC activation regulates several platelet responses including GPIIb-IIIa activation and secretion, and megakaryocyte differentiation. Deficiency of a human platelet PKC isozyme (PKC-) has been described in a patient previously reported with mucocutaneous bleeding manifestations, mild thrombocytopenia, and markedly abnormal platelet aggregation (including in primary wave) and dense granule secretion in response to multiple agonists. Phosphorylation of pleckstrin and myosin light chain was diminished in the patient’s platelets on activation with PAF and thrombin. Signal transduction–dependent activation of GPIIb-IIIa was impaired on stimulation with receptor-mediated agonists. This subject has a heterozygous mutation in a transcription factor, RUNX1 (see also the section on Platelet Function Abnormalities Associated with Transcription Factor Deficiencies). The platelets were specifically deficient in one PKC isozyme, PKC-. This deficiency provides one cogent explanation for the impaired protein phosphorylation and abnormal aggregation and secretion.
Protein phosphorylation by other kinases (tyrosine kinases) is important in platelet signaling events. In thrombasthenia and the Scott syndrome, tyrosine phosphorylation of several proteins is impaired on platelet activation. In these disorders, this defect is likely secondary to the primary abnormality in the GPIIb-IIIa complex and in phospholipids scrambling, respectively. Interestingly, in patients with the thrombocytopenia with absent radii (TAR) syndrome, thrombopoietin-induced tyrosine phosphorylation is markedly abnormal.
Abnormalities in Arachidonic Acid Pathways and Thromboxane Production
Thromboxane A2 production forms an important positive feedback, enhancing the overall activation process. In the absence of TxA 2 synthesis, dense granule secretion is decreased following stimulation of PRP with ADP, epinephrine, and low concentrations of collagen and thrombin. In general, most patients with defects in TxA 2 production have had mild to moderate bleeding manifestations.
Defects in the liberation of arachidonic acid from phospholipids
Mobilization of free arachidonic acid from membrane-bound phospholipids by phospholipase A 2 is the initial and rate-limiting step in TxA 2 synthesis. Defects in this process have been reported in some patients. In 4 such patients, aggregation and secretion were abnormal, and TxA2 production was diminished during stimulation with ADP and thrombin, but was normal with free arachidonic acid. In 3 H-arachidonic acid–labeled platelets, thrombin-induced mobilization of free arachidonic acid from phospholipids was impaired in these patients. Subsequent studies in one of the patients showed that the platelet PLA 2 levels (both membrane and cytosolic) were normal but agonist-induced Ca 2+ mobilization was impaired due to a platelet Gαq deficiency. Other reports have also documented patients with an impaired release of arachidonic acid. An inherited deficiency in cytosolic phospholipase A 2 , the principal enzyme that regulates the release of arachidonic acid, has been reported in a patient with recurrent small intestinal ulceration, markedly decreased synthesis of thromboxane, 12-HETE and leukotriene B 4 , and platelet dysfunction. This patient had 2 heterozygous single base-pair mutations in the PLA 2 coding region, leading to S111P and R485H substitutions.
Deficiencies of cyclooxygenase and thromboxane synthetase
In 1975, Malmsten and colleagues reported platelet cyclooxygenase deficiency in a patient with a mild bleeding disorder and impaired aggregation responses to ADP, epinephrine, collagen, and arachidonic acid, but with normal response to PGG2. Subsequently, several other patients have been described with a similar defect in TxA 2 synthesis. Studies using a radioimmunoassay found normal levels of cyclooxygenase in 5 of 6 patients suspected to have a deficiency, suggesting that these patients have a functionally abnormal molecule. Three patients have been described with impaired platelet responses and markedly decreased ability to convert arachidonic acid, but not of PGH 2 , to TxA 2 . The investigators demonstrated decreased platelet cyclooxygenase-1 levels in 2 patients and normal levels in the third; levels of thromboxane synthase were normal in all 3. Two patients have been described with thromboxane synthetase deficiency.
Related posts:
 Harnessing the Platelet Signaling Network to Produce an Optimal Hemostatic Response
Genetic Dissection of Platelet Function in Health and Disease Using Systems Biology
Congenital Thrombocytopenia
Drug-induced Immune Thrombocytopenia
Drug-induced Immune Thrombocytopenia
Diagnosis and Management of Heparin-Induced Thrombocytopenia
Harnessing the Platelet Signaling Network to Produce an Optimal Hemostatic Response
Genetic Dissection of Platelet Function in Health and Disease Using Systems Biology
Congenital Thrombocytopenia
Drug-induced Immune Thrombocytopenia
Drug-induced Immune Thrombocytopenia
Diagnosis and Management of Heparin-Induced Thrombocytopenia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


