Although much remains to be learned about the heritable factors involved, enormous strides have been made in the past two decades in understanding inherited susceptibility to breast cancer. These advances are based on the discovery and characterization of a number of high-risk, relatively uncommon genes responsible for the clustering of breast cancer in certain families. More recently, a large number of common variants having a modest effect on individual risk have been defined by the use of genome-wide association studies. As clinical utility is currently largely restricted to high-risk genes, this chapter will focus largely on this category but in the future it seems possible that low-risk common variants will also be utilized to inform risk and management of breast cancer. Other relevant information can be found in Chapter 17.
One measure of familial clustering is the familial relative risk (FRR) which is defined as the ratio of the risk of breast cancer for a relative of an affected individual to that of the general population. Multiple observations including simulation and twin studies suggest that the FRR for breast cancer largely reflects the genetic influence on the disease.
Although genetics are clearly important, there is a tendency to assume that familial clustering of disease invariably results from inherited predisposition. However, other explanations for familial clustering of breast cancer should be considered including (a) geographically limited environmental exposure to carcinogens, which might affect an extended family living in close proximity; (b) culturally motivated behavior that alters risk factor profile, such as age at first live birth; and (c) socioeconomic influences that, for example, might result in differing dietary exposures. In addition, multiple, complex inherited genetic factors likely influence the extent to which a risk factor for breast cancer plays a role in any one individual; such modifying effects are likely to be shared among genetically related members of an extended family.
HISTORICAL EPIDEMIOLOGIC STUDIES OF FAMILIAL BREAST CANCER
The first attempts to determine the influence of family history on breast cancer risk were published in the first half of the twentieth century (1, 2). Although many of these studies have methodological flaws, they consistently demonstrated a twofold to threefold increase in breast cancer risk in mothers and sisters of patients with breast cancer. The first large population-based study to estimate breast cancer risk associated with a family history was conducted in Sweden and involved 2,660 women (3). Within this study cohort, women with an affected relative had an increased breast cancer risk of 1.7 compared to those without. Anderson (4) suggested that a small subset of families with a very high risk of developing breast cancer due to a single genetic defect might be obscured in studies in which most breast cancer cases were multifactorial in origin. By 1980, a significant body of evidence had accumulated supporting the presence of inherited factors responsible for familial clustering of breast cancer, and efforts shifted to determining the inheritance pattern of breast cancer within these families. In 1984, Williams and Anderson (5) provided the first evidence for an autosomal dominant breast cancer susceptibility gene with age-related penetrance finding supported by Newman et al. (6) in 1988.
MODE OF INHERITANCE
To date, all studies of inherited susceptibility to breast cancer suggest that breast cancer susceptibility is transmitted in an autosomal dominant mendelian fashion, and the identification of an increasing number of genes has born out this modeling (7, 8) (Table 16-1). With a pattern of autosomal dominant inheritance, an individual can have one of three possible genotypes: carrier of two nonmutant alleles (homozygous normal), or carrier of one (heterozygous) or two (homozygous) mutant alleles. The actual risk of developing breast cancer in a mutation carrier is based on the penetrance of the gene. Penetrance is the likelihood that the effect (phenotype) of a mutation (genotype) will become clinically apparent. Individuals carrying two copies of an autosomal dominant disease-related gene are rare, partly because of the relative rarity of heterozygotes and partly because of the potential for a lethal defect in a homozygous affected fetus. However, biallelic (homozygous) deleterious mutations in BRCA2 have been reported in patients with Fanconi anemia type D1, a rare recessive disorder characterized by leukemia and birth defects (9). Finally, there are several reports of individuals who have both BRCA1 and BRCA2 mutations (10). Anecdotal observations suggest that these women develop more frequent and earlier cancers than single mutation carriers, but the number of such individuals identified is too small for definitive studies.
TABLE 16-1 Allele Frequency and Effect Sizes Associates with High-, Moderate-, and Intermediate-Penetrance Variants
Locus
Genes in/Near Region
Variant
MAF
RR
High-penetrance mutations
17q21
BRCA1
0.0006
5-45
13q12.3
BRCA2
0.001
9-21
17p13.1
TP53
rare
2-10
10q23.3
PTEN
rare
2-10
19p13.3
STK11
rare
2-10
16q22.1
CDH1
rare
2-10
Moderate-penetrance variants
11q22.3
ATM
0.003
2-3
22q12.1
CHEK2
0.004
2-3
17q22-q24
BRIP1
0.001
2-3
16p12.1
PALB2
rare
2-4
Low-penetrance variants
10q26
FGFR2
rs 2981582
0.38
1.26
16q12
TOX3
rs 3803662
0.25
1.20
5q11
MAP3K1
rs 889312
0.28
1.13
8q24
FAM84B/c-MYC
rs 13281615
0.40
1.08
11p15
LSP1
rs 3817198
0.30
1.07
3p24
NEK10/SLC4A7
rs 4973768
0.46
1.11
17q23.2
COX11
rs 6504950
0.27
0.95
10p14
CASP8 (D302H)
rs 1045485
0.13
0.88
2q35
TNP1/GFBP5/IGFBP2/TNS1
rs 13387042
0.52
1.12
1p11.2
NOTCH2/FCGR1B
rs 11249433
0.40
1.14
14q24.1
RAD51L1
rs 999737
0.24
0.84
5p12
MRPS30/FGFR10
rs10941679
0.26
1.19
6q25.1
ESR1
rs 2046210
0.35
1.29
MAF, minor allele frequency from European populations; RR, relative risk. From Mavaddat N, Antoniou AC, Easton DF, et al. Genetic susceptibility to breast cancer. Mol Oncol 2010;4:174-191.
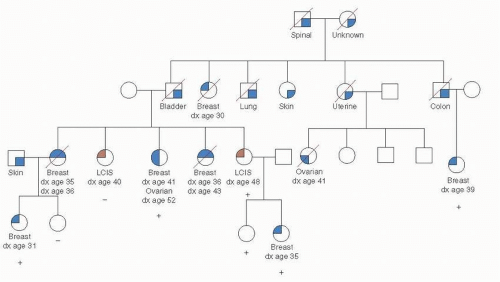
There is a 50% chance that an individual offspring will inherit a mutant copy of any given gene from a heterozygous parent. Therefore, on average, 50% of the related individuals in a family carry the mutant gene being transmitted. If the penetrance of the gene is high, the pedigree pattern for an autosomal dominant disease is quite striking, with vertical inheritance and half the children of an affected parent also being affected, whereas none of the offspring of a homozygous normal parent are affected. This pedigree pattern also presupposes a low risk in the general population, which is not the case for breast cancer. As a result, breast cancer in women from families that have a known BRCA1 mutation but who do not themselves carry the mutation is not uncommon. Such women are termed phenocopies, because they have the phenotype associated with the gene mutation but are noncarriers. This situation is illustrated in the pedigree shown in Figure 16-1, a typical pedigree of a family known to carry a mutation in BRCA1. As long as the gene being examined is not on the X or Y sex-related chromosomes, the sex of the carrier is irrelevant. However, in the case of autosomal dominant inheritance of breast cancer, significant sex-related differences in the penetrance of mutations exist. Therefore, although mutations occur equally in male and female populations, breast cancer is much more common in women with BRCA1 or BRCA2 mutations than in men, but male breast cancer is part of the spectrum of both BRCA1 and BRCA2.
FIGURE 16-1 A kindred with a BRCA1 mutation. □, Unaffected cancers are indicated with dark shading of symbols;(+), known BRCA1 mutation carriers;(−), individuals who tested negative; all others are untested. Deceased individuals are indicated with a diagonal line through the symbol. One family member with lobular carcinoma in situ (LCIS) tested positive, and the other tested negative, consistent with previous reports suggesting LCIS is not a component of BRCA1-related cancer susceptibility.
TUMOR SUPPRESSOR GENES
Two fundamental types of genetic alterations responsible for the development of the malignant phenotype are found in cancer cells: (a) activation of protooncogenes producing a “gain of function” in the affected cell and (b) inactivation of tumor suppressor genes producing a “loss of function” in the cell. Some tumor suppressor genes are important in cell-cycle regulation, normally functioning as checks on cell growth; others are critical elements in the cellular response to DNA damage, preventing the propagation of mutations in other critical genes. Mutated tumor suppressor genes lose these regulatory functions, leading to malignant transformation. However, because all individuals are born with two alleles of every gene, an explanation was needed for the development of cancer in large numbers of individuals who had only a single inherited mutation in a tumor suppressor gene. In 1971, Knudson (11) put forward the “two-hit hypothesis,” suggesting that cancer arises as a result of two genetic events occurring in the same cell, inactivating both copies of a given tumor suppressor gene. In the case of sporadic cancer (i.e., cancer occurring in women without a family history of the disease), the likelihood that two events would occur in the same gene in the same cell is quite low. However, individuals from “cancer families” inherit an inactivating mutation in one allele of the implicated tumor suppressor gene in all cells (i.e., a germline mutation); therefore, only one somatic (noninherited) event is required to inactivate the single remaining copy, making the development of cancer a much more common event than in individuals born without the “first hit.” Of particular relevance to breast cancer are the tumor suppressor genes TP53, BRCA1, and BRCA2.
HEREDITARY BREAST CANCER SYNDROMES
The study of clinical syndromes that include an increased incidence of breast cancer has provided insight into the mechanisms by which genetic mutations result in the development of cancer. The most frequently identified pedigrees contain site-specific breast cancer (i.e., breast cancer in these families is not found in association with inherited susceptibility to other cancers, such as ovarian) and are thought to represent the effect of a single genetic abnormality; BRCA1 and BRCA2 are the best studied examples. Breast cancer also has been noted to occur in association with other cancers. The occurrence of breast cancer in association with diverse childhood neoplasms in the Li-Fraumeni/SBLA (soft-tissue and bony sarcomas, brain tumors, leukemias, and adrenocortical carcinomas) (12) syndrome and the association between breast and ovarian cancer represent some of the most intensively studied examples. An elevated frequency of breast cancer may occur in patients with hereditary syndromes that include nonmalignant manifestations as well, such as Cowden’s disease and Muir-Torre syndrome (13, 14 and 15). An increasing number of moderate-risk genes—ATM, CHEK2, PALB2, and BRIP1—are being identified that lead to an increased risk of cancer of twofold to fourfold (8). Finally, numerous common variants (population frequency 5% to 50%) in genes, which cause a very modest (1.1-1.5 fold) elevation in risk, are just starting to become part of the landscape of breast cancer susceptibility (8) (Table 16-1).
BRCA1 and BRCA2
In 1990, chromosome 17q21 was identified as the location of a susceptibility gene for early onset breast cancer, now termed BRCA1 (16). Shortly thereafter, linkage between the genetic marker D17S74 on 17q21 and the appearance of ovarian cancer in several large kindreds was also demonstrated (17). Initial estimates suggested that BRCA1 mutations were responsible for more than 90% of breast cancer cases in families with apparent autosomal dominant transmission of breast cancer and at least one case of ovarian cancer, and 45% of cases in families with breast cancer only. However, the percentage of site-specific breast cancer cases attributed to BRCA1 mutations rose to almost 70% if the median age at onset of breast cancer in the families was younger than 45 years (18), demonstrating the critical importance of the characteristics of a family to the likelihood that a BRCA1 mutation will be detectable. The BRCA1 gene was identified in 1994 (19) and encodes a novel protein now known to be important in the cellular response to DNA damage (20).
Initial progress toward the identification of a second breast cancer susceptibility gene came from a linkage analysis of 22 families with multiple cases of early onset female breast cancer and at least one case of male breast cancer. Linkage between male breast cancer and polymorphic genetic markers on chromosome 13q12-13 identified the BRCA2 locus (21). In 1995, the partial sequence of BRCA2 and six germline mutations that truncated the putative BRCA2 protein were identified (22). Shortly thereafter, the complete structure of the BRCA2 gene was published (23).
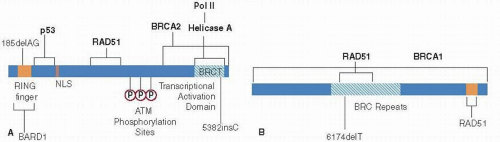
FIGURE 16-2(A) Functional domains of BRCA1. An idiogram of the 220-kd BRCA1 protein depicting known functional domains. Domains are shown as filled areas within the diagram. The two common mutations found in the Ashkenazi Jewish population (185delAG and 5382insC) are indicated. (B) Functional domains of BRCA2. The carboxy terminal RAD51 binding site, and the central RAD51 binding BRC repeats are as depicted. The common mutation (6174delT) found in the Ashkenazi Jewish population is indicated (51, 52).
BRCA1 is composed of 24 exons (coding regions) and is translated into a protein consisting of 1,863 amino acids (Fig. 16-2A). The coding region of BRCA2 is 11.2 kb in length and is made up of 26 exons that produce a protein of 3,418 amino acids. The size of these genes is important from a clinical standpoint in the context of genetic testing, because this has made screening for mutations technically demanding and costly. Furthermore, the BRCA1 gene contains a large number of repetitive elements that facilitate the generation of large deletions and duplications. For example, diseaseassociated deletions account for 36% of BRCA1 mutations in the Netherlands (24). However, the use of modern nextgeneration DNA sequencing methodologies are already overcoming these technical and cost issues.
More than 500 coding region sequence variations have been detected in BRCA1 and 250 in BRCA2. A listing and description of most known mutations is available on the Breast Cancer Information Core (BIC) website (research. nhgri.nih.gov/bic/). Several similarities between BRCA1 and BRCA2 are apparent. No mutation hot spots in either have been detected. Most unequivocally confirmed mutations reported to date are truncating mutations, adding little in the way of clues for defining functional regions. Finally, few mutations have been identified in either gene in sporadic breast cancers. However, it has been suggested that the pathways in which the BRCA1 and BRCA2 proteins act may be disrupted in sporadic cancer, a phenotype that has been termed “BRCAness” (25).
Estimates of BRCA1 and BRCA2 mutation prevalence in unselected patients with breast cancer are in the range of 2% to 3%. In a large population-based study of white and black cases (n = 1,628) and controls (n = 674) in North America for ages 35 to 64, BRCA1 mutations were detected in 2.4% of cases and 0.04% of controls while BRCA2 mutations were detected in 2.3% of cases and 0.4% of controls. BRCA1 mutations were more common in white (2.9%) than black (1.4%) of cases, while BRCA2 mutations were slightly more frequent in black (2.6%) than white (2.1%) cases (7). In families identified through clinics treating high-risk breast cancer, BRCA1 and BRCA2 mutations are found in up to 55% of families with both breast and ovarian cancer and up to 75% of families with both breast and ovarian cancer in the same individual, underscoring the importance of the family history in determining the likelihood that a BRCA1 mutation is present (26). Population-based DNA sequencing studies, now feasible due to new technology, should give much more accurate estimates of mutation prevalence.
Population Genetics of BRCA1 and BRCA2
The population genetics of BRCA1 and BRCA2 reflect several basic evolutionary principles. Each gene has undergone multiple independent mutations and these mutations have migrated with the populations in which they originally occurred. Certain “founder mutations” are known to exist in BRCA1 and BRCA2, which have occurred in specific ethnic populations many generations in the past. They persist because the development of disease usually occurs after childbearing age, so individuals carrying these mutations are able to pass them on to subsequent generations with little impact of the mutated alleles on survival of the species.
Founder mutations have been identified in a number of populations. A comprehensive review by Szabo and King (27) reveals the similarities and differences in mutation rate, penetrance, and nature of the mutations among various population groups. The proportion of high-risk families with breast or ovarian cancer appears to vary widely by population group. Mutations in BRCA1 are most common in Russia (79% of families with breast and/or ovarian cancer), as compared to Israel (47% of families) and Italy (29%). BRCA2 mutations appear to be more common than BRCA1 mutations only in Iceland, where a single mutation accounts for virtually all of the BRCA2-associated breast and ovarian cancer cases (28).
BRCA1 and BRCA2 mutations among the Ashkenazi Jewish population are among the most intensively researched, as the presence of founder mutations facilitates these studies. The two Ashkenazi Jewish founder mutations in BRCA1 are 185delAG and 5382insC, occurring in 1 in 8 and 1 in 12 individuals of Ashkenazi descent, respectively (29, 30). One of these two mutations, 6174delT in BRCA2, occurs in more than 2% of the Ashkenazi Jewish population. When compared to the estimated frequency of BRCA1 mutations in an unselected white population of about 0.1% (31), this finding suggested the presence of a “founder effect” in the Ashkenazi Jewish population, documented with haplotype studies (32). Analysis of germline BRCA1 mutations in several cohorts of Jewish women suggests that more than 20% of Jewish women developing breast cancer before age 40 carry the 185delAG mutation (33). Even more strikingly, estimates suggest that 30% to 60% of all Ashkenazi Jewish women with ovarian cancer carry one of the BRCA1 or BRCA2 founder mutations (34). Up to 90% of mutations identified in women of Ashkenazi Jewish descent are one of the three founder mutations, although other BRCA1 and BRCA2 mutations have also been detected (35). Based on these data, individuals of Ashkenazi descent choosing to undergo genetic testing should first be tested for the three Ashkenazi Jewish founder mutations. Full sequencing can be reserved for those individuals at particularly high risk of having a BRCA1 or BRCA2 mutation.
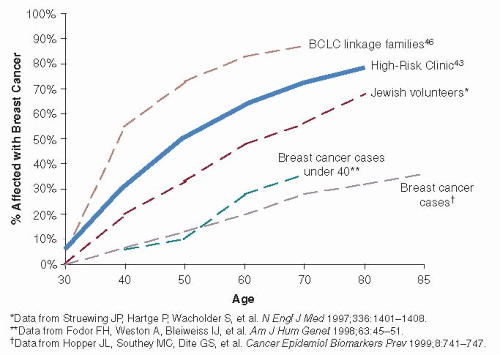
FIGURE 16-3 Breast cancer risk estimates associated with BRCA1 mutations vary depending on sample ascertainment. Breast cancer risks (penetrance) will be highest in families selected to have multiple affected family members for use in linkage studies (27) and lowest in population-based ascertainments (29). Sample sets collected in breast cancer risk evaluation clinics would be expected to be intermediate between these two ascertainments; recent data have confirmed that hypothesis (30). An ascertainment of Ashkenazi Jewish volunteers also falls between high- and low-risk penetrance estimates, again because this sample is likely a mix of population-based ascertainment and individuals who volunteer because they were aware of a strong family history (31).
Though still limited, data are now available on the prevalence of BRCA1 and BRCA2 mutations in some nonwhite populations. Interestingly, many BRCA1 and BRCA2 mutations in African Americans appear unique to this ethnic/racial group (36) and in addition, genetic testing for BRCA1 and BRCA2 mutations in the African American population is complicated by a high rate of variants of unknown significance. More data are also becoming available from the Hispanic population, with similar features predicting pathogenic mutations (37). Comprehensive data from other ethnic groups or geographic areas are lacking.
Cancer Risks for BRCA1 and BRCA2 Mutation Carriers
Cancer risk estimates for BRCA1 and BRCA2 mutation carriers have been controversial (38). Estimates based on the highly penetrant families used to find these genes are high (as they were selected to be), likely due to coexistent genetic and environmental modifiers that may increase the risk of disease. However, in studies of lower-risk cohorts, such as population-based studies or cohorts of women with breast cancer unselected for family history, the lifetime risk of breast cancer was much lower (39). For this reason, the estimation of the risk of breast cancer in BRCA1 mutation carriers has been variable with an estimate of pooled data of 65% (40) (Fig. 16-3). Estimates of contralateral breast cancer occurrence are as high as 60% (41). Cumulative risk of ovarian cancer in BRCA1 carriers has been reported to be between 27% (40) and 45% (42, 43), and there is also a significantly increased risk of fallopian tube cancer and reports of an increase in uterine and cervical cancer, stomach cancer, a twofold to threefold increase in pancreatic cancer, a possible twofold increase in colon cancer, and a 17-fold risk of testicular cancer (43, 44); however, these risks have not been consistently seen across studies. Prostate cancer risk does not appear increased, although the disease may occur at an earlier age. Male breast cancer is also seen in association with BRCA1 mutations (43).
BRCA2 has a cancer risk profile similar, but not identical, to BRCA1. Lifetime breast cancer risk for BRCA2 mutation carriers is estimated to be 45% to 84%, with lifetime ovarian cancer risk in the range of 10% to 20% (40, 45) and BRCA2 mutations are associated with a 6% lifetime risk of male breast cancer (7). Male BRCA2 mutation carriers have an increased risk of prostate cancer. Pancreatic cancer also is associated with BRCA2 mutations (46), with an RR of 3.5. The incidence of BRCA2 germline mutations in patients with familial pancreatic cancer (two first-degree relatives with pancreatic cancer) may be as high as 20% (47). BRCA2 mutation carriers also appear to have an increased risk of stomach cancer (RR = 2.6), gallbladder and bile duct cancers (RR = 5.0), and malignant melanoma (RR = 2.6) (46).
Modifiers of BRCA1 and BRCA2 Mutations
Although germline mutations in BRCA1 and BRCA2 confer a high risk of breast cancer, a great deal of variability in cancer risk among individuals both between and within families has been observed. The discovery of environmental or genetic factors that modify the penetrance of BRCA1 and BRCA2 mutations may clarify our understanding of their mechanism of action and provide additional information with which to counsel individuals with BRCA1 and BRCA2 mutations. Furthermore, factors that affect familial breast cancer risk in the general population could presumably affect breast cancer risk in BRCA1 and BRCA2 mutation carriers. By far the most important modifiers identified for BRCA1 and BRCA2 mutation carriers are prophylactic oophorectomy and the use of tamoxifen for chemoprevention. Prophylactic oophorectomy decreases the risk of ovarian cancer by 95% but importantly also decreases the risk of breast cancer by 50% (48, 49). The magnitude of the benefit of oophorectomy (and estrogen deprivation) on breast cancer risk is seen in both in BRCA1 and BRCA2 mutation carriers, despite that 90% of BRCA1-related breast cancers are ER negative. In retrospective studies, tamoxifen has been shown to decrease the risk of contralateral breast tumors by 50% in both BRCA1 and BRCA2 mutation carriers (50). To date, many reproductive factors have been examined as modifiers in BRCA1 and BRCA2 mutation carriers, including parity, age at first pregnancy, oral contraceptive use, and tubal ligation (51). Of all these, perhaps the most clinically relevant are the protective effects of oral contraceptives on ovarian cancer risk.
Only gold members can continue reading. Log In or Register to continue
 Breast Cancer Screening
Breast Cancer Screening
 Ductal Carcinoma In Situ and Microinvasive Carcinoma
Ductal Carcinoma In Situ and Microinvasive Carcinoma
 Adjuvant Systemic Therapy: Endocrine Therapy
Adjuvant Systemic Therapy: Endocrine Therapy
 Preoperative Endocrine Therapy for Operable Breast Cancer
Preoperative Endocrine Therapy for Operable Breast Cancer



