11.1.2 Inheritance and Clinical Picture
A large epidemiological study conducted in a young normal population in Italy in 1987 showed a prevalence of VWD of 1 % or 1 in 100 [2]. In a study in which the United States Nationwide Inpatient Sample (NIS) for the years 2000–2003 was queried for pregnancy-related discharges, women with a diagnosis of VWD were compared with women without VWD; the frequency of a diagnosis of VWD among women giving birth was 0.024 % or 1 in 4,000 [3]. A study of symptomatic patients at hemostasis centers found an incidence of 0.002–0.01 % or 1 in 10,000 [4].
There are explanations for some of these discrepancies – the level of VWF in a young population tends to be lower, and it has been found that the level of VWF increases by 15 % each decade [5]. National Indicator set data are derived from discharge summaries, and there is a strong possibility of errors in coding toward under-diagnosis. Furthermore, many patients remain undiagnosed or are seen outside hemostasis centers, particularly in the USA [3]. It has also been pointed out that the wide spectrum of clinical and laboratory manifestations and the lack of strong penetrance mean that making the diagnosis can be very difficult [6].
Thus, VWD is the most common inherited bleeding disorder, and a low von Willebrand factor (VWF) may be found with a frequency of 1 % in young women [2].
11.1.3 Classification of VWD
von Willebrand disease has been classified into three main types [7].
Type 1 is a quantitative deficiency of VWF with a mild to moderate VWF level, 15–50 IU/dL, and normal VWF multimer structure. It occurs in approximately 85 % of cases.
Type 2 is a qualitative deficiency occurring in 15 % of cases and is further subdivided – type 2A, loss of high- and intermediate-weight multimers; type 2B, loss of high molecular weight multimers and often associated with thrombocytopenia; and type 2M, normal VWF multimers and poor binding to platelets. Type 2N VW involves a mutation in the VWF protein which leads to decreased binding of FVIII with resultant FVIII deficiency, and is often misdiagnosed as hemophilia A.
Type 3 is a severe quantitative deficiency with undetectable VWF protein. It is rare, occurring at a frequency of one in a million in the UK population. However, in those populations where first cousin marriage is practised, it is more common [8].
11.1.4 Prepregnancy Counseling
Type 1 and 2 von Willebrand disease is inherited as an autosomal dominant condition with variable inheritance; therefore for type 1 and 2 VWD, there is a 50 % risk of a mother transmitting VWD to her child. However, type 3 VWD is an autosomal recessive disorder, and an affected individual is either homozygote or compound heterozygote. Thus, when a child with type 3 VWD has already been born in a family, the risk of a subsequent child being affected is 25 %. This is common in first cousin marriages; the parents may request antenatal diagnosis, but this should be planned in advance to allow the causative mutation to be identified [9].
11.1.5 Antenatal Management
The physiological response to pregnancy is an increase in FVIII and VWF; thus, most women with VWD do not bleed excessively during pregnancy (Fig. 11.2) [10, 11].
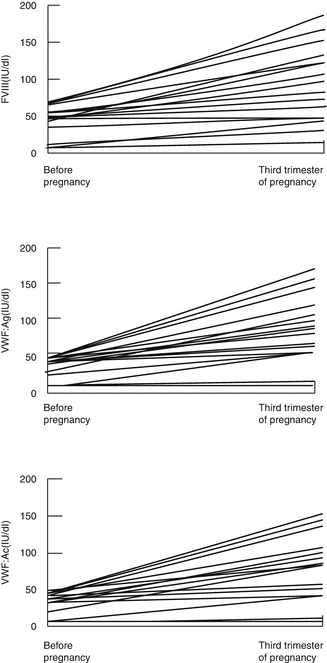
Most women with type 1 VWD achieve levels in the normal nonpregnant range by the third trimester [10]. In a series of 24 pregnancies in 24 women with VWD studied retrospectively, it was found that the FVIIIC and VWF:Ag rose above baseline levels by 1.5 times in most cases. However, some women who are severely affected with type 1 VWD may fail to achieve normal VWF levels by the third trimester, and a baseline level of <15 IU/dL was predictive of a third trimester level <50 IU/dL [12].
In type 2 VWD, although FVIII and VWF:Ag levels increase, there is minimal or no increase in VWF:AC levels [12]. In type 2N VWD, the FVIII level remains low because of impaired binding by VWF. In type 2B VWD, thrombocytopenia may worsen in pregnancy because the increase in intermediate multimers induces platelet aggregation [13].
Regular monitoring of VWF:Ag and VWF:AC together with FVIIIC is recommended at booking, 28 and 34 weeks ‘gestation, and prior to invasive procedures [9]. The platelet count should be monitored in those with type 2B VWD [13]. If the VWF:AC does not reach 50 IU/dL by the third trimester, prophylaxis with clotting factor concentrate to cover delivery should be considered [9, 15].
11.1.6 Miscarriage
The spontaneous miscarriage rate has been reported as 20 % [16]. In a review of 84 pregnancies in women with VWD during the years 1980–1996, it was found that 33 % presented with vaginal bleeding during the first trimester and there was an overall spontaneous miscarriage rate of 21 % [10]. A miscarriage rate of 21 % was reported in another study of pregnant women with VWD [17]. Thus, although women with VWD have a higher rate of bleeding during the first trimester, there is not an increased rate of miscarriage.
Spontaneous miscarriage or elective termination in women with VWD is associated with an increased risk of bleeding complications [10, 13, 17]. In one study, transfusion was required for bleeding in association with 10 % of spontaneous miscarriages or elective abortions, and intermittent bleeding occurred for 2 weeks after miscarriage in 30 % of cases [10]. Most miscarriages occur during the first trimester before the VWF: AC has increased substantially [12]. It is therefore recommended that women with VWD who present with spontaneous miscarriage or who elect for termination of pregnancy should have the VWF:AC measured and prophylactic treatment given if the VWF:AC is <50 IU/dL [9].
11.1.7 Treatment
11.1.7.1 DDAVP
The use of DDAVP in pregnancy remains controversial. A survey in the USA showed that 50 and 30 % of hematologists used intravenous and intranasal DDAVP, respectively, for postpartum hemorrhage (PPH) in type 1 VWD but 31 % considered pregnancy as a contraindication [18]. There are reports of the use of DDAVP to cover chorionic villus sampling or amniocentesis [19], to cover 52 deliveries and 20 Cesarean sections in women with a low third trimester VWF [20], and to cover mothers with VWD after cutting the cord. [21] In a systematic review of the literature including 168 pregnancies with VWD, DDAVP was shown to be effective in preventing and reducing bleeding complications associated with pregnancy and childbirth without any complications [22]. The evidence suggests that DDAVP is not contraindicated in uncomplicated pregnancy.
11.1.7.2 Clotting Factor Concentrate Containing VWF
Virally inactive plasma-derived concentrate containing VWF is the treatment of choice for those women who are unresponsive to DDAVP. If the FVIIIC and/ or VWF:AC is <50 IU/dL, a prophylactic infusion should be started at the onset of labor and a VWF:AC >50 IU/dL maintained for at least 3 days post vaginal delivery and 5 days post-Cesarean section [9]. In type 2 N VWD, recombinant FVIII has been successfully used [23]. In type 3 VWD, concentrate is always required to cover delivery because the VWF level does not increase in pregnancy [14].
11.1.8 Regional Analgesia and Anesthesia
Several case reports have described women with VWD who received epidural anaesthesia without bleeding complications [24–26]. In a series, eight women with VWD received regional anesthesia during labour and delivery without bleeding complications and only one woman received prophylactic therapy as the clotting factor levels were >50 IU/dL in the other cases [27]. Epidural anesthesia may be considered for use in the majority of women with type 1 VWD whose levels have risen to >50 IU dL. However, the decision on its use needs to be made jointly by an experienced anesthetist, obstetrician and hematologist after consideration is given to hemostatic concerns such as the degree of correction of the plasma FVIII:C and VWF levels, possible degree of residual platelet impairment, possible rate of postpartum decline of VWF and the consequent risks of bleeding/spinal hematoma. The risks of an epidural or spinal anesthetic for Cesarean section should be balanced against the risk of a general anesthetic. In all cases the epidural should be inserted by an experienced anesthetist. Epidural anesthesia is generally not recommended for use in type 2 or 3 VWD [28].
11.1.9 Postpartum Management
There is an increased risk of both primary (>500 mL blood loss in the first 24 h after delivery) and secondary (excessive bleeding from 24 h to 6 weeks postpartum) PPH in women with VWD. This is due to a fall in VWF and FVIIIC postnatally [11] (Fig. 11.3). Three series including 51 women and 92 deliveries showed a primary PPH rate of 16–29 % and a secondary PPH rate of 20–29 % [10, 12, 29]. All women with VWD should therefore be advised to have active management of the third stage of labor; this involves administration of an oxytocic drug with the birth of the baby, early cord clamping, and controlled cord traction. The VWF level should be checked postdelivery particularly in those with a low pre-delivery baseline. The risk of PPH is higher in types 2 and 3 VWD, and in these women, factor replacement should be given during labour and delivery to maintain VWF >50 IU/dL for at least 3 days post vaginal delivery or 5 days post-Cesarean section in addition to tranexamic acid 1 g 6 hourly until the lochia is stopped. The fall in VWF postdelivery can be very variable – there are anecdotal reports of a fall from 41 to 9 IU/dL over the course of a week [12] and a fall of 50 % within 24 h of delivery [30]. One study found that the average time of presentation of secondary PPH in women with VWD is 16 days postdelivery [31] and therefore there is a need for observation and possibly prophylaxis for several weeks postpartum. Prolonged and/or intermittent secondary PPH has also been reported in VWD [10, 12]. In a recent study, the duration of lochia was significantly longer in women with VWD compared to those with no bleeding disorders [32].
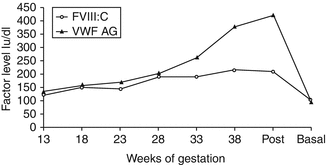
Fig. 11.3
The levels of FVIII and VWF:Ag during pregnancy and postdelivery (From Stirling et al. [11])
11.2 Factor XI Deficiency
11.2.1 Inheritance and Clinical Picture
The management of pregnancy and delivery for women with Factor XI (FXI) deficiency is difficult because the bleeding tendency is so unpredictable. This deficiency was first described in 1953 [33] and was termed hemophilia C. Although the inheritance was initially thought to be autosomal dominant [34], a study in 1961 showed autosomal recessive inheritance [35]. Factor XI deficiency was found to be very common in Jews where the frequency of the heterozygous state among Ashkenazi Jews was found to be 8 % [36].
Individuals with severe FXI deficiency, FXI below 15–20 IU/dL, are at risk of bleeding with surgery and trauma, but some individuals with this severe deficiency do not have a bleeding tendency [37]. The bleeding may be inconsistent within a particular family and is not related to the factor level, in contrast to hemophilias A and B [38, 39]. Studies of the relationship of the FXIC level and the bleeding tendency have also shown that individuals with a level between 50 and 70 IU/dL may have a bleeding tendency [37, 38] (Fig. 11.4). Thus, it is important to take a good bleeding history and to test for additional coexisting abnormalities of VWF and platelet function [40].
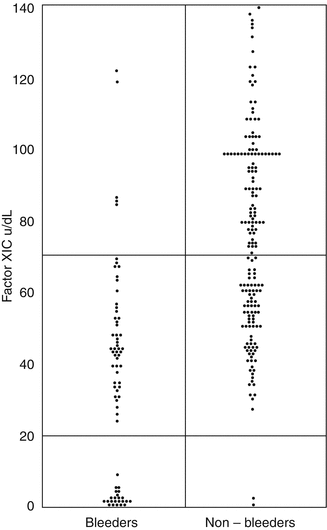
Fig. 11.4
The relationship between FXIC level and bleeding tendency. Pooled data from two UK studies [33, 34] – 249 individuals from 54 families transmitting factor XI deficiency. Of 128 heterozygotes, 45 (35 %) are bleeders. The upper horizontal line defines the lower limit of the normal range, and the lower line cut-off between severe and partial deficiency (From Bolton-Maggs [37])
11.2.2 Treatment of FXI Deficiency
11.2.2.1 Fresh Frozen Plasma (FFP)
FFP was the mainstay of treatment until FXI plasma-derived concentrate became available. This was used in Rosenthal’s original patients [33]. FFP has been made safer with respect to possible viral transmission either by pooled sol-vent detergent treated plasma or single donor units treated with methylene blue [42]. However, in the UK, there remains a concern about possible transmission of abnormal prions following the large epidemic of BSE in cattle and ingestion by potential plasma donors [43].
11.2.2.2 Plasma-Derived FXI Concentrate
A plasma-derived FXI concentrate derived from German plasma is available in the UK [42]. However, there have been concerns about potential thrombogenicity [44], therefore the dose used should not raise the plasma level above 70 IU/dL, and the maximum dose should be 30 IU/dL. The use of FXI concentrate should be limited to those with severe deficiency (<20 IU/dL) or in mild deficiency (20–70 IU/dL) where there is a clear history of bleeding [42].
11.2.2.3 Tranexamic Acid
Tranexamic acid alone has been demonstrated to be sufficient to cover dental extractions in people with severe FXI deficiency [45]. It can be used in those with milder FXI deficiency (20–70 IU/dL) where bleeding is difficult to predict. However, tranexamic acid should not be used concurrently with FXI concentrate because of the potential thrombotic risk [42].
11.2.2.4 Recombinant Factor VIIa
Recombinant VIIa (rVIIa) has been used to cover surgery in FXI deficiency in order to overcome the potential risk of transfusion-transmitted infection. The dose used was 90 mcg/kg intravenously 2 h for the first 24 h, and 4 hourly for the second 24 h [46]. Low doses (30 mcg/kg) have recently been used and shown to effectively induce hemostasis in patients with FXI deficiency undergoing surgery [47]. Due to the unpredictable length of labor and short half-life of rFVIIa, its use is not practical to cover labor, but it has been used successfully to cover elective Cesarean section.
11.2.3 Genetic Counseling and Prenatal Diagnosis
Prenatal diagnosis should be offered to patients where there is a risk of severe deficiency [9]. A partner of a female with FXI deficiency should be offered screening with a FXIC assay.
11.2.4 Antenatal Management
Factor XI levels usually remain constant during pregnancy [10, 50, 51]. A study in London followed 61 pregnancies in 30 women, 2 with severe deficiency (FXI 2–8 IU/dL) and 28 with partial deficiency (FXI median 51 IU/dL, range 34–65 IU/dL). The median (range) prepregnancy and third trimester FXI levels were 55 (34–65) and 54 (37–75) IU/dL, respectively, in women with a partial deficiency, and 2 and 8 and 4 and 13 IU/dL in the two women with severe deficiency. Overall, the FXI levels did not change significantly during pregnancy (p = 0.09) [50]. Routine monitoring of FXI levels should ideally be carried out at booking, 28 and 34 weeks. The rate of miscarriage does not seem to be increased in these women [51], but they may be at increased risk of hemorrhage following miscarriage or termination of pregnancy, especially if there is a bleeding history [10, 51]. Therefore, consideration of prophylaxis with FXI concentrate or virally inactivated FFP should be given to those with severe FXI deficiency or a bleeding history for invasive procedures, such as CVS or amniocentesis, and termination of pregnancy. Probable “nonbleeders” can be managed expectantly, with treatment available [9].
11.2.5 Intrapartum Management
The risk of PPH can be reduced by active management of the third stage of labor and prophylactic use. FXI concentrate or solvent detergent treated fresh frozen plasma (SD-FFP) if FXI concentrate is not available [10, 52–54]. In severe deficiency (FXI < 20 IU/dL), especially those with a positive bleeding history, FXI concentrate in a dose such that peak levels do not exceed 70 IU/dL (NR 70–150 IU/dL) is recommended, regardless of the mode of delivery, to be given at the onset of labour, or prior to induction of labour or Cesarean section. For women with partial deficiency and a bleeding history, or no previous hemostatic challenge, tranexamic acid 1G 6–8 hourly during labor and postdelivery for 7 days is recommended. A “wait and watch” policy is advised for those with partial deficiency and no bleeding history despite hemostatic challenge [9]. The decision to give prophylactic cover for Cesarean section in women with partial deficiency should be individualized and dependent on bleeding history and type of anesthetic or analgesia [9, 50, 55].
11.2.6 Regional Analgesia and Anesthesia
Epidural anesthesia has been carried out in FXI-deficient women without complication [10, 51, 53]. A total of 60 women with inherited bleeding disorders have been identified in the English literature describing the use of regional block during labor and delivery, and there were no reported complications [50, 55]. Epidural or spinal block should be covered with tranexamic acid or FXI concentrate depending on the mother’s factor FXI level and her bleeding tendency – FFP contains a variable quantity of FXI and is not recommended [9]. Consideration can be given to the use of rFVIIa [56].
11.2.7 Postpartum Management
Women with FXI deficiency are at increased risk of postpartum hemorrhage [10, 38, 51]. A recent series documented obstetric outcome in 61 pregnancies in 30 women with FXI deficiency over 10 years (1997–2006) [51]. The rate of PPH was 11 % for both primary and secondary PPH but only 3 of the 10 PPH occurred after prophylaxis with factor XI concentrate or tranexamic acid was given – clearly prophylactic hemostatic support reduces PPH as the rate of primary PPH and secondary PPH has previously been reported as 16 and 24 %, respectively, in untreated patients (Fig. 11.5, [10, 50, 55]). Prophylactic tranexamic acid in a dose of 1G 6 hourly should be considered for 3 days post vaginal delivery partum or 5 days following Cesarean section. Since the half-life of FXI is 52 h [55], subsequent doses of FXI concentrate are rarely needed [9].
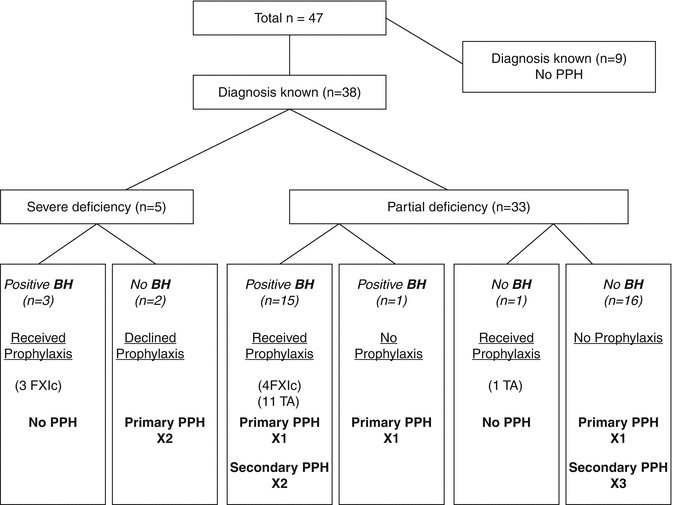
Fig. 11.5
The use of intrapartum prophylaxis and postpartum hemorrhage in women with FXI deficiency (From Chi et al. [50]). BH bleeding history, TA tranexamic acid
11.3 Hemophilia A and Hemophilia B
11.3.1 Inheritance
Hemophilia A and B are sex-linked recessive disorders with an incidence in male births of 1 in 5,000 and 1 in 30,000, respectively. The daughter of a male with hemophilia is an obligate carrier, and a carrier has a 50 % chance of passing the gene defect to her children – there is a 50 % chance of having an affected son and a 50 % chance of having a daughter who is a carrier (Fig. 11.6). Carriers may have a reduced clotting factor level but a wide range of values, 22–116 IU/dL, has been reported [57] because of the phenomenon of lyonization where there is random inactivation of one of the two X chromosomes [58]. Where there is extreme lyonization, the clotting factor level may be very low with an increased risk of bleeding. However, there is also an increased bleeding risk at mildly reduced clotting factor levels [59]. Management of carriers of hemophilia requires a multidisciplinary approach, and ideally they should be managed in a combined clinic with both expertise and facilities to provide a management plan [60].
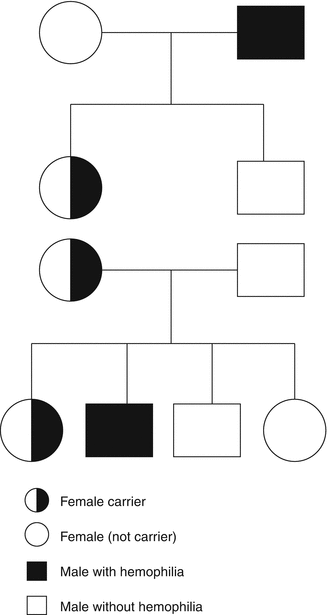
Fig. 11.6
The inheritance of hemophilia
11.3.2 Genetic Counseling and Carrier Detection
The purpose of genetic counseling is to provide the potential carrier and her parents or partner with the information required to reach a decision about carrier testing and prenatal diagnosis and to provide support during the process [61]. Genetic counseling for hemophilia includes discussion of the medical condition – the inheritance and treatment, personal and relationship concerns related to hemophilia, and beliefs and wishes of the person discussing possible inheritance, as well as others who might be affected [61]. A framework for genetic service provision has been provided as a guideline by the UK Haemophilia Centre Doctors’ Organisation (UKHCDO) [62] (Table 11.1). The first step in carrier diagnosis is the family tree – the daughter of a man with hemophilia is an obligate carrier and her sons have a 50 % chance of having hemophilia and her daughters have a 50 % chance of being a carrier (Fig. 11.6). Within a family where an index patient with hemophilia has been identified, there may be many females at risk of being carriers. However, newly diagnosed cases of hemophilia may be sporadic – one study reported this as being 50 % of newly diagnosed cases [63]. However, in a mother of a sporadic case of hemophilia, there is the possibility of mosaicism – a mixture of normal and mutation carrying cells. Carriership should be established before pregnancy and before prenatal diagnosis.
Table 11.1
Counseling and consent for genetic testing in hemophilia
Establish that hemophilia is present in the family and determine its type and severity |
Establish family pedigree (tree) to identify possible or definite (obligate) carriers |
Provide a full explanation of the potential clinical effects of being a hemophilia carrier or affected male |
Provide a full explanation of mode of inheritance of hemophilia
Related posts: Systemic Thromboembolism in Pregnancy: Heritable and Acquired Thrombophilias
Inherited Bleeding Disorders in Pregnancy: Rare Coagulation Factor Defects
Inherited Bleeding Disorders in Pregnancy: Platelet Defects
The Neonate Systemic Thromboembolism in Pregnancy: Heritable and Acquired Thrombophilias
Inherited Bleeding Disorders in Pregnancy: Rare Coagulation Factor Defects
Inherited Bleeding Disorders in Pregnancy: Platelet Defects
The Neonate
 Paroxysmal Nocturnal Hemoglobinuria in Pregnancy Paroxysmal Nocturnal Hemoglobinuria in Pregnancy
 Antithrombotic Therapy for Cardiac Disorders in Pregnancy Antithrombotic Therapy for Cardiac Disorders in Pregnancy
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|