Fig. 19.1
Pathophysiology of PNH
19.3 Pregnancy and PNH
Pregnancy and childbirth pose particular clinical challenges in women with PNH. Information about the management of PNH during pregnancy is limited to case reports and small studies [15, 25–27]. Pregnancy in PNH patients is associated with high rates of maternal and fetal morbidity and mortality, and there is further increased risk during the post-partum period [14, 27, 28]. Maternal mortality has been reported to be as high at 12–21 % [14, 27], although this is probably an overestimate due to over-reporting of eventful cases. However, there is also a significant risk of complications such as stroke, which, although not always fatal, can lead to permanent deterioration in health. The main causes of death have been thromboembolism, more often in unusual sites such as hepatic and cerebral veins, and infections. The perinatal mortality rate has been reported to be 7–9 %, with prematurity the main risk to the baby [14, 27]. Stillbirth and miscarriage is also common and over 45 % of pregnancies in women with PNH result in either spontaneous miscarriages or early termination [12]. It has been reported that only half of the pregnancies progress to term. Major maternal complications requiring hospitalization and/or admission to an intensive care unit can occur in 16 % of women, with the highest incidence during the post-partum period [14]. Women with PNH may present for the first time during pregnancy. A retrospective review demonstrated that the diagnosis of PNH in women was made during pregnancy in 25 % of cases [27]. As for most other rare diseases, the primary step in the successful management of PNH is to consider the diagnosis as it can be mistaken for other conditions in pregnancy. PNH should be considered as a diagnosis in pregnant women presenting with unexplained anemia and/or thrombocytopenia with intravascular hemolysis as well as thrombosis in unusual sites [29, 30]. Worsening anemia due to increased intravascular hemolysis and thrombocytopenia are often more severe during pregnancy than in the non-pregnant state, resulting in an increased red cell transfusion requirement throughout pregnancy [14, 27, 28, 31]. The platelet count is often less than 50 × 109/L [27], which makes the antithrombotic management of thrombosis more challenging during pregnancy.
19.3.1 Thrombosis During Pregnancy and the Postpartum Period
Normal pregnancy is accompanied by increased concentrations of fibrinogen, FVII, FVIII, FX, and von Willebrand factor, particularly marked in the third trimester [32–35]. The active form of protein S is decreased during pregnancy, secondary to increased levels of its binding protein, the complement component C4b [32, 36]. Plasminogen activator inhibitor type 1 (PAI-1) levels are also increased [37]. All of these changes contribute to the hypercoagulable state seen in pregnancy and in women with PNH contribute to the further increased risk of thrombosis. Venous thromboembolism (VTE), comprising DVT and PE, is a leading cause of morbidity and mortality during pregnancy in the western world. The risk of pregnancy associated VTE has been estimated from 0.5 to 2 per 1,000 pregnancies [38–40]. The risk of developing DVT and PE in pregnancy is five to tenfold higher than in non-pregnant women of child-bearing age [41]. The incidence of postpartum VTE is more common than antepartum and 20 times more likely following Cesarean section than vaginal birth [38]. VTE accounts for 0.79 deaths per 100,000 pregnancies [42]. Compared to VTE, arterial thrombosis is far less common in pregnancy, with the reported incidence of stroke and myocardial infarction being 0.18 and 0.1 per 1,000 births, respectively [43].
In PNH, there is an increased risk of both venous and arterial thromboembolism, with the incidence of VTE in patients with PNH around 40 % [2]. VTE is the main cause of mortality in PNH, accounting for 40–67 % of PNH-related deaths whose cause is known [44]. As in the non-pregnant state, as well as lower limb DVT and PE, the hepatic and cerebral veins are the sites most commonly affected with thrombosis in PNH [2, 45]. The mechanism of thrombosis in PNH is not fully understood and multiple mechanisms have been proposed [46], including endothelial damage caused by the free hemoglobin released from intravascular hemolysis, platelet activation and microparticle formation [47]. It has been shown that there is increased procoagulant and fibrinolytic activity in patients with PNH, suggesting increased fibrin generation [48]. Several defects have been observed in the fibrinolytic system, including a deficiency of urokinase-type plasminogen activator receptor on PNH granulocytes, and increased plasma levels of soluble urokinase-type plasminogen activator receptors [49]. Furthermore, it has been shown that patients with a large PNH clone have a greater risk of developing thrombosis, suggesting a direct relationship between the PNH clone size and risk of thrombosis [44, 50]. Thrombosis may occur despite anticoagulation, and in one reported case a pregnant woman developed Budd–Chiari syndrome whilst on therapeutic dose anticoagulation, as well as bone marrow failure [51]. Another case report (Case Study 1 below) describes a woman with PNH who appears to have developed sagittal sinus thrombosis despite prophylactic dose low molecular weight heparin (LMWH) [30].
19.3.2 Renal Disease
Although renal failure itself is not a common problem during pregnancy in women with PNH, it contributes to 8–18 % of PNH related deaths [52]. Renal damage in PNH is associated with chronic hemosiderosis and/or microvascular thrombosis, renal vein thrombosis and recurrent urinary tract infections [53, 54]. Evidence of renal damage (on renal biopsy, by using imaging techniques or at post-mortem examination) can be seen in almost all PNH patients [52, 55–60]. Repeated exposure to cell-free hemoglobin release from intravascular hemolysis causes hemosiderin accumulation in renal tubules, tubulo-interstitial inflammation and renal damage [58].
Free hemoglobin scavenges NO leading to its depletion. NO is of critical importance in the regulation of vascular tone with the greatest effect on the afferent renal arterioles [61]. Impairment of renal blood flow because of alteration of vascular tone due to lack of NO can have a direct effect on the glomerular filtration rate and renal plasma flow [61–64]. During pregnancy, there is a risk of renal vein thrombosis leading to acute renal failure and increased hemolysis, which in turn leads to release of more free hemoglobin. Hemoglobinuria is associated with recurrent urinary tract infections, predominantly in women with PNH [54], with a further increased risk during pregnancy. Good hydration and careful monitoring of blood pressure, in addition to other management strategies, is critical during pregnancy and will be discussed later in the chapter.
19.3.3 Bone Marrow Failure and Increased Hemolysis During Pregnancy
Physiological hematological changes in pregnancy include dilutional anemia, relative neutropenia and gestational thrombocytopenia [65]. In addition, iron deficiency anemia is common during pregnancy and folate deficiency can also occur. It may sometimes be difficult to distinguish the manifestations of PNH from those of normal pregnancy and from other pathological conditions which can occur during pregnancy such as HELLP syndrome (Hemolysis, Elevated Liver enzymes and Low Platelets), preeclampsia, gestational thrombocytopenia and immune thrombocytopenia [66]. HELLP syndrome, in particular, is often difficult to distinguish from a PNH crisis during pregnancy as both conditions share common features. Normally, HELLP syndrome develops in the third trimester of pregnancy and clinical features can include right upper quadrant or epigastric abdominal pain and tenderness, nausea, and vomiting. Laboratory abnormalities include thrombocytopenia <100 × 109/L, low haptoglobin, elevated lactate dehydrogenase (LDH) and indirect (unconjugated) bilirubin [67]. These laboratory findings are almost always present in PNH too, and patients with PNH commonly experience abdominal pain, which can also be due to PNH-related hepatic vein thrombosis. HELLP syndrome is typically associated with microangiopathic changes (i.e. red cell fragmentation in association with thrombocytopenia) on the blood film that are not usually seen in PNH. Although hypertension is not a feature of PNH, it may accompany a PNH crisis or can occur due to renal damage, which is common in patients with PNH. Furthermore, a woman with PNH can present with preeclampsia/HELLP syndrome during pregnancy [27]. The severity of hemolysis increases during pregnancy especially in later pregnancy. Women with PNH have been noted to experience hemoglobinuria prior to their next dose of eculizumab even if they had well controlled hemolysis on the standard fortnightly doses of eculizumab for years prior to becoming pregnant [15].
Both anemia and thrombocytopenia are common in women with PNH during pregnancy, and in the pre-eculizumab era often necessitated red cell and platelet transfusion [25, 27]. A series of 27 pregnancies in 22 women with PNH from France reported that the majority (95 %) developed at least minor complications, mainly cytopenias, during pregnancy, requiring transfusion in more than half, while two women developed aplastic anemia (AA) during pregnancy [25]. In this case series, it was observed that in one of the cases with AA, spontaneous improvement of hematological parameters occurred after delivery with only persistent asymptomatic hemolysis. In a review of 20 published reports describing the outcome of 33 pregnant women with PNH [27], there were 73 and 27 % event rates for anemia and thrombocytopenia, respectively. The hemoglobin levels were typically below 80 g/L and, of those who developed anemia, 83 % required red cell transfusion, often more than once. Platelet counts were typically less than 50 × 109/L and some women developed major bleeding events, whilst one of these women developed a major VTE episode with a platelet count of 10 × 109/L. In this report the authors were unable to assess the rate of pancytopenia.
19.4 Diagnosis of PNH
PNH may be diagnosed during or outside of pregnancy. A comprehensive clinical history is critical and PNH should be suspected and investigated in patients presenting with any of the following: hemoglobinuria; direct antiglobulin test (DAT) negative hemolytic anemia; thrombosis in unusual sites such as Budd-Chiari syndrome, other intra-abdominal sites (e.g. mesenteric or portal veins), cerebral veins, dermal veins; aplastic anemia (such patients should be screened for PNH at diagnosis and annually even in the absence of evidence of intravascular hemolysis), refractory anemia/myelodysplastic syndrome (MDS); and episodic dysphagia or abdominal pain with evidence of intravascular hemolysis [68].
Laboratory investigations for PNH include:
Full blood count (FBC), reticulocytes and blood film, particularly to check for red cell fragmentation
LDH, total bilirubin, direct bilirubin, haptoglobin, serum B12 and folate levels, urinary hemosiderin, serum iron studies (iron concentration, total iron binding capacity, transferrin saturation index, and ferritin concentration, with C-reactive protein estimation as ferritin is an acute phase reactant)
Direct antiglobulin test (DAT); blood group and red cell alloantibody screen
Flow-cytometric analysis of GPI-anchored proteins
Bone-marrow diagnostics including cytology, cytogenetics and histology, in patients with significant cytopenia of such an extent that PNH is suspected in the context of another hematological disease (especially aplastic anemia or MDS).
Flow cytometric analysis using antibodies directed against GPI-AP is the most sensitive and informative assay available for diagnosis of PNH [68–70]. For initial studies, quantitation of at least 2 GPI-APs is recommended to exclude the possibility that the clinical process is a consequence of an inherited, isolated deficiency of a single GPI-AP [71, 72]. For patients with established, stable disease, annual analysis of peripheral blood GPI-AP expression is recommended; however, a change in clinical parameters (worsening or more frequent hemolysis, or a thromboembolic event) warrants immediate re-evaluation [68]. The flow cytometric analysis can both determine the percentage of cells that are abnormal and identify discrete populations with different degrees of deficiency (particularly on red cells; Fig. 19.2). Red cells with complete deficiency of GPI-APs are called PNH III, those with subtotal deficiency (usually around 10 % of normal expression) PNH II, and those with normal expression PNH I (Fig. 19.2). In order to obtain accurate information about the percentage of GPI-AP–deficient erythrocytes, flow cytometric analysis should be performed prior to red cell transfusion; in a transfused patient, the analysis should be deferred for at least 4 weeks or longer if clinically safe to do so. This is because the flow cytometric histogram will be affected by the normal (transfused) red cells by increasing the proportion of red cells expressing CD55 and CD59, although it is unlikely to obscure the diagnosis of PNH. Analysis of expression of GPI-AP on granulocytes provides additional clinically useful information such as aiding the decision to start prophylactic anticoagulation as the size of the PNH clone has a direct relationship to the risk of thrombosis, as mentioned previously. The current recommendation is to commence prophylactic anticoagulation if the PNH granulocytes clone is >50 % [50]. Unlike GPI-AP–deficient red cells, the life span of PNH granulocytes is normal [73, 74], hence the proportion of abnormal granulocytes more accurately reflects the PNH clone size and is unaffected by red cell transfusion [68] (Fig. 19.3).
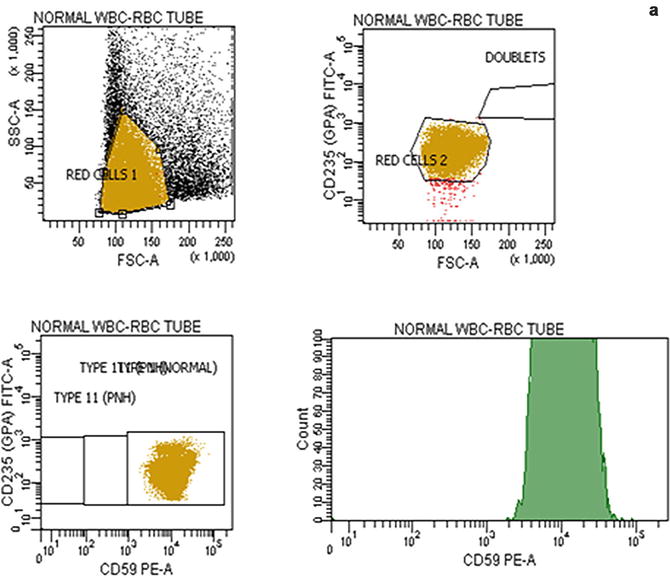
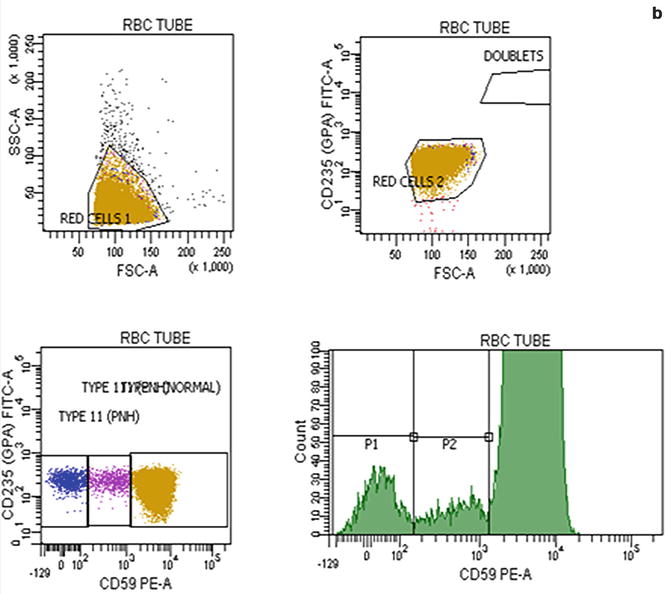
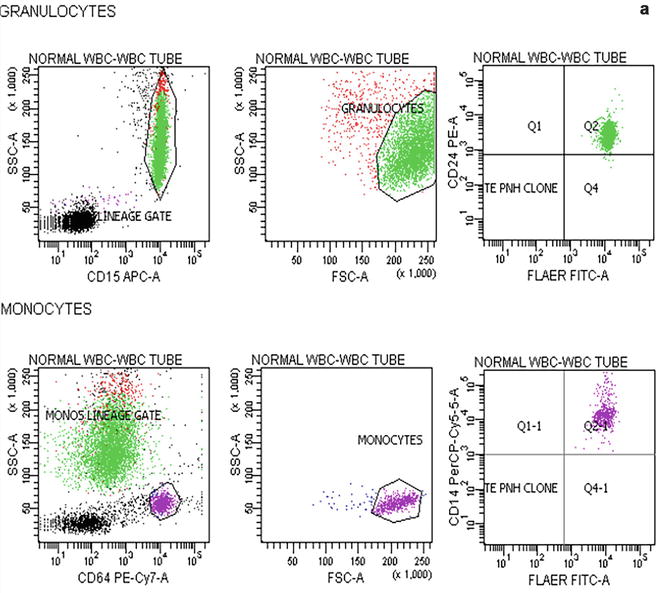
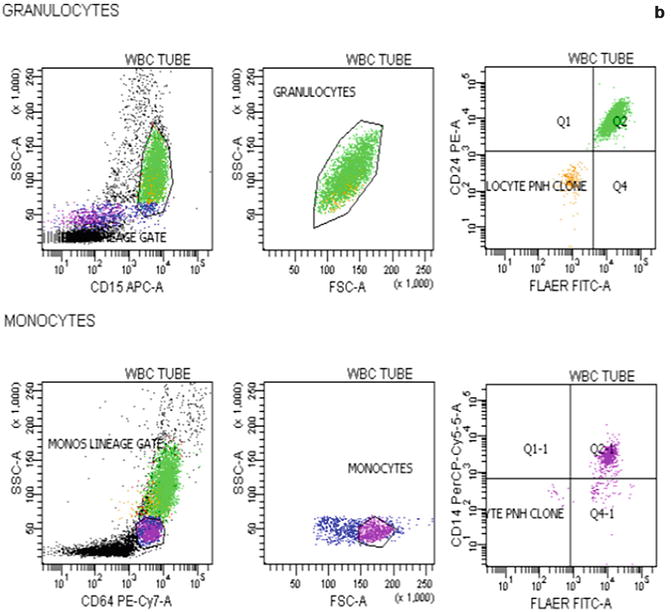
Fig. 19.2
Flow cytometric analysis of red cells in a normal individual (a) and in a patient with PNH (b) using anti-CD59. A normal individual’s histogram analysis of CD59 expression shows a single population of cells positive for CD59. In a patient with PNH, the histogram demonstrates that, in addition to a major population of CD59 positive normal red cells, there are two populations of red cells deficient in CD59. These are PNH red cells
Fig. 19.3
Flow cytometric analysis of granulocytes in a normal individual (a) and in a patient with PNH (b) using anti-CD15 and anti-CD64. In the patient with PNH there are two populations of cells that are outside the normal CD15 and CD64 gated area. These are PNH granulocytes
19.5 Management of PNH During Pregnancy
The management of women with PNH during pregnancy and postpartum is challenging and should be undertaken in a multidisciplinary high risk clinic setting, with specialist hematological and obstetric input. It is important to refer these patients to a tertiary centre with experience in managing patients with PNH for further advice and management. Women with PNH should receive pre-pregnancy counselling, preferably at an early stage of management. The patient’s age, overall health, previous history of thrombosis, degree of hemolysis and bone marrow failure, particularly thrombocytopenia, the size of the PNH clone, other co-existing medical conditions and the degree of personal support should all be taken into account [68]. The management of patients with hemolytic or thrombotic PNH (generally those with a PNH granulocyte clone of over 50 %) is described below and in Table 19.1. Patients with predominantly aplastic anemia and those with small PNH clones should be monitored closely but do not necessarily require anticoagulation and/or eculizumab. An individualised management plan should be established and documented for each patient.
Table 19.1
The management of pregnancy in patients with haemolytic and/or thrombotic PNH
The management of women with PNH during pregnancy requires close monitoring and specialist hematological and obstetric input |
An individualised management plan should be established and documented for each patient |
Eculizumab should be administered during and immediately after pregnancy, with the dose increased should there be breakthrough haemolysis during pregnancy |
Split therapeutic dose low molecular weight heparin (LMWH) should be commenced as soon as pregnancy is confirmed, and continued throughout pregnancy and for at least 6 weeks postpartum |
Red cell and platelet transfusions should be administered as required |
Delivery should be planned in advance and undertaken in a hospital with expertise in obstetric high risk patients and specialist haematological input |
Normal vaginal delivery is recommended unless there is an obstetric reason for instrumental delivery or cesarean section |
Anticoagulation should be restarted as soon as is feasible post-delivery, in order to minimise the additional risk of thrombosis during this period of high risk due to excessive complement activation resulting from tissue injury |
In the case of significant bleeding during pregnancy or at delivery, fresh frozen plasma (FFP) should be used with great caution, as it contains a large amount of complement, which markedly increases the risk of thrombosis and intravascular hemolysis. If its use is essential, FFP must be covered with eculizumab. Prothrombin complex concentrate may be considered but should be used with caution due to the risk of thrombosis |
19.5.1 Supportive Therapy and Anticoagulation
Close monitoring of the pregnancy, both clinical and laboratory, with regular ultrasound scans to monitor the fetus is important. Both anemia and thrombocytopenia frequently worsen during pregnancy. Red cell and platelet transfusion should be administered as clinically required. In the case of significant bleeding during pregnancy or at delivery, fresh frozen plasma (FFP) should be used with great caution, as it contains a large amount of complement, which markedly increases the risk of thrombosis and intravascular hemolysis [66]. If its use is essential, FFP must be covered with eculizumab [66]. Prothrombin complex concentrate may be considered but should be used with caution due to the risk of thrombosis [15]. Iron and folic acid supplements should be given to all women with PNH throughout pregnancy. Although the role of prophylactic anticoagulation during pregnancy has not been studied systematically, given the high risk of thrombosis during pregnancy, which carries significant risk of morbidity and mortality, the current approach is to start anticoagulation with therapeutic dose LMWH as soon as the pregnancy is confirmed and continue throughout pregnancy and for at least 6 weeks post-partum [68]. It is important that women who are already on anticoagulation for previous thrombosis with warfarin or other vitamin K antagonists (VKA) should be switched to split therapeutic dose LMWH (i.e. 12 hourly) as soon as pregnancy is confirmed, and before 6 weeks’ gestation, due to the risk of teratogenicity in the first trimester. Regular monitoring of the blood count as well as the reticulocyte count and LDH is required to assess the degree of intravascular hemolysis, and liver function and renal function tests should be undertaken. Close monitoring of the platelet count should be undertaken, and prophylactic platelet transfusion should be administered if required to avoid bleeding, particularly with concurrent use of anticoagulation. Anti-Xa levels are useful to monitor the anticoagulant intensity of LMWH. The lower anti-Xa peak levels achieved with split dose LMWH minimise the risk of bleeding should the platelet count fall.
19.5.2 Use of Eculizumab During Pregnancy
Eculizumab [75], a humanized monoclonal antibody, inhibits the terminal complement cascade by binding uniquely to human complement protein C5, thereby inhibiting the formation of pro-inflammatory, prothrombotic C5a and C5b, with subsequent inhibition of assembly of the membrane attack complex [11, 76, 77] (Fig. 19.4). Following a series of multinational clinical trials demonstrating the efficacy and safety of eculizumab in patients with PNH [11, 44, 78, 79], it was approved for use in PNH by the United States Food and Drug Administration (FDA) in March 2007 and by the European Medicines Agency (EMA) in June 2007. Eculizumab is well tolerated, leads to a rapid and clinically significant reduction in intravascular hemolysis, thus providing substantial clinical benefit [77] and improvement in patients’ quality of life [11]. Eculizumab is given initially as 600 mg once weekly by intravenous infusion (IVI) for 4 weeks followed by 900 mg 1 week later, and thereafter 900 mg every 14 days [11, 78]. However, under certain circumstances a dose adjustment will be necessary because of the occurrence of breakthrough hemolysis during pregnancy (e.g. maintenance therapy with 900 mg weekly rather than fortnightly). As terminal complement deficiency is associated with an increased risk of infections with encapsulated organisms [80], mainly Neisseria meningitides, all patients must be must be given meningococcal vaccine. The risk of meningococcal infection following vaccination is low and it has been reported as an infection rate of 0.42 per 100 patient-years [77]. The current practice in the UK is to administer tetravalent meningococcal vaccine on the day the patient starts eculizumab (given the potential for complement activation and complications with vaccination) and to cover the first 2 weeks of therapy with an effective dose of ciprofloxacin followed by penicillin V 500 mg bd indefinitely. In the last 4 years since the UK policy was changed there have been no episodes of meningococcal infection in over 150 patients treated.
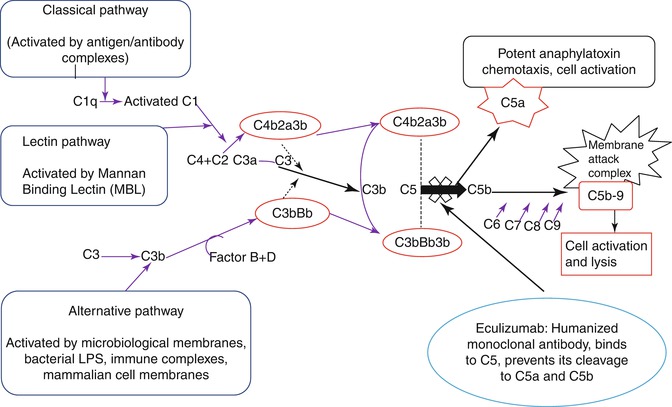
Fig. 19.4
Eculizumab a humanized monoclonal antibody, inhibits the terminal complement cascade by binding uniquely to human complement protein C5, thereby inhibiting the formation of pro-inflammatory, prothrombotic C5a, and C5b, with subsequent inhibition of assembly of the membrane attack complex
The observation that eculizumab was effective and well tolerated in red cell transfusion-dependent patients with PNH were demonstrated to be also applicable to a broader PNH patient population, which included those with minimal transfusion requirements or with thrombocytopenia, in the phase 3 open-label non-placebo-controlled SHEPHERD (Safety and Efficacy of the Terminal Complement Inhibitor Eculizumab in Patients with Paroxysmal Nocturnal Hemoglobinuria) study. This international multicenter study included 97 patients from 33 sites. Treatment with eculizumab resulted in an 87 % reduction in hemolysis. It also led to an improvement in anemia despite a 52 % reduction in transfusions, with 49 (51 %) of patients not requiring transfusion for the study period of 52 weeks. The improvements in hemolysis, fatigue, and transfusion requirements with eculizumab were independent of baseline levels of hemolysis and degree of thrombocytopenia. Quality of life measures were also broadly improved with eculizumab treatment [81]. The long-term safety and efficacy of eculizumab was investigated in 195 patients over 66 months, and found to have a substantial impact on the symptoms and complications of PNH and result in a significant improvement in patient survival [77].
Related posts:
 Systemic Thromboembolism in Pregnancy: Heritable and Acquired Thrombophilias
Anticoagulants and Antiplatelet Agents in Pregnancy
Acquired Bleeding Disorders in Pregnancy: Obstetric Hemorrhage
Thrombocytopenia in Pregnancy: Gestational Thrombocytopenia and Immune Thrombocytopenic Purpura
Systemic Thromboembolism in Pregnancy: Venous Thromboembolism
Thrombotic and Hemostatic Aspects of Assisted Conception
Systemic Thromboembolism in Pregnancy: Heritable and Acquired Thrombophilias
Anticoagulants and Antiplatelet Agents in Pregnancy
Acquired Bleeding Disorders in Pregnancy: Obstetric Hemorrhage
Thrombocytopenia in Pregnancy: Gestational Thrombocytopenia and Immune Thrombocytopenic Purpura
Systemic Thromboembolism in Pregnancy: Venous Thromboembolism
Thrombotic and Hemostatic Aspects of Assisted Conception
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


