Infectious Gastroenteritis
L. Clifford McDonald
Benjamin A. Lopman
INTRODUCTION
Infectious gastroenteritis (IG) is manifest most commonly by diarrhea and variable degrees of nausea and vomiting. As a healthcare-associated infection (HAI), IG is unique from other HAIs such as surgical site infections, urinary tract infections, pneumonia, and bloodstream infection, with no direct role for invasive devices or procedures in pathogenesis. In addition, the main manifestations are more commonly due to noninfectious rather than infectious causes. Diarrhea, defined as three or more unformed stools in a 24-hour period, is the principal symptom of IG with nausea and vomiting playing a more prominent secondary role in certain etiologies such as norovirus. Healthcare-associated diarrhea, defined as diarrhea with onset ≥3 days after admission is common (1). In one recent prevalence study, healthcare-associated diarrhea was found in 12% of patients overall and in 27% of patients who had been hospitalized for >3 weeks (2).
Instead of IG, medications and enteral feedings are the most common cause of healthcare-associated diarrhea (1). Among causes of IG, pathogens may be categorized on the basis of whether symptoms are associated with antibiotics or not. Antibiotics, because they disrupt the lower intestinal microbiota, are responsible for 25% of all episodes of healthcare-associated diarrhea even without a recognized pathogen playing a distinct role. When IG is found in antibiotic-associated diarrhea (AAD), it is most often due to Clostridium difficile, the most common healthcare-associated IG and recognized cause of AAD. The next most common IG cause of AAD is Klebsiella oxytoca, which causes a distinctly hemorrhagic colitis and commonly resolves with cessation of the antibiotics. Clostridium perfringens and Staphylococcus aureus are two even less prominent IG causes of ADD. Meanwhile, the most prominent IG cause of healthcare-associated diarrhea not associated with antibiotics is norovirus. The primary focus of this chapter is on the two most well-recognized healthcare-associated IGs, C. difficile and norovirus infections, but also includes a brief discussion of other viral causes of IG that may be transmitted in healthcare settings.
CLOSTRIDIUM DIFFICILE INFECTION (CDI)
THE ORGANISM AND ITS VIRULENCE FACTORS
C. difficile is an anaerobic, gram-positive, spore-forming bacillus. It was first isolated from a healthy infant in 1935 and named difficile because, as an obligate anaerobe, it can be difficult to cultivate in the laboratory (3). It was first associated with human disease in the late 1970s when Koch’s postulates were fulfilled, establishing it as the cause of pseudomembranous colitis (4,5). Pseudomembranous colitis, meanwhile, had been first described in the late 1800s (6), but became much more common in the antibiotic era and recognized as a particular threat following the use of clindamycin in the 1970s to the point that it was commonly known as “clindamycin colitis” (7). In retrospect, this was likely due to early emergence of highly clindamycin-resistant strains of C. difficile, foreshadowing how years later, acquired resistance to commonly used antibiotics would facilitate the emergence of epidemic strains (8,9).
The main virulence factors of C. difficile are the two large clostridial toxins A and B (10,11). Both toxins have high sequence homology, suggesting evolutionary origins via a gene replication event. Both have carboxy-terminal binding domains, amino-terminal biologically active domains, and a small hydrophobic intermediate domain, possibly active in translocation. The main biologic activity is via glycosylation of Rho proteins, disrupting the cell cytoskeleton. Whereas toxin A targets only cells of the gastrointestinal endothelium, toxin B targets a broad range of cells. While it was initially believed that toxin A was the main virulence factor in humans, over and above toxin B, this was called into question in the early 1990s with the emergence of toxin A− B+ strains that could cause even severe disease (12). More recent laboratory work with isogenic C. difficile isolates suggests that toxin B may be more important or that, at the very least, either toxin individually can cause disease (13,14). Both toxin genes and surrounding regulatory genes are located in a 19.6-Kb region of the C. difficile genome known as the pathogenicity locus (11). In addition to toxins A and B, a third toxin, known as binary toxin, was historically found in <10% of isolates. The gene encoding binary toxin is not located in the pathogenicity locus, and although its exact role in pathogenesis is not known, it has increased in prevalence as it is present in hypervirulent strains that have recently emerged (15).
PATHOGENESIS AND CLINICAL PRESENTATION
C. difficile is not part of the normal lower-intestinal microbiota, but is acquired following transmission via the fecal-oral route from other patients (16). The spore is the transmissible form that is ingested and, owing to its relative acid resistance, passes easily through the stomach and into the small bowel where it germinates into the active growing vegetative form and then passes into the large intestine. Following acquisition, the human host may either become asymptomatically colonized, develop infection, or resist either colonization or infection. The main human host defense mechanisms that determine outcome following transmission are an intact lower-intestinal microbiome (i.e., the collective genome of the microbiota
reflecting both strain and species composition) and humoral immunity or responsiveness (17). An intact intestinal microbiome prevents both colonization and infection; antibiotic exposures that commonly perturb the microbiome are the principal modifiable risk factor for both infection and colonization. Asymptomatic colonization occurs in two major patient groups: infants who have not yet established a mature microbiome and adults who have a perturbed microbiome following antibiotic exposure. Colonization rates may easily exceed 50% in either of these groups (18,19,20), depending upon the opportunity for transmission from other colonized or infected individuals (Figure 34.1). Meanwhile, rates of detectable C. difficile carriage in healthy adults who have not recently received antibiotics are <5%, and even among these persons the organism may be only passing through the gastrointestinal tract following environmental exposure (21,22,23).
reflecting both strain and species composition) and humoral immunity or responsiveness (17). An intact intestinal microbiome prevents both colonization and infection; antibiotic exposures that commonly perturb the microbiome are the principal modifiable risk factor for both infection and colonization. Asymptomatic colonization occurs in two major patient groups: infants who have not yet established a mature microbiome and adults who have a perturbed microbiome following antibiotic exposure. Colonization rates may easily exceed 50% in either of these groups (18,19,20), depending upon the opportunity for transmission from other colonized or infected individuals (Figure 34.1). Meanwhile, rates of detectable C. difficile carriage in healthy adults who have not recently received antibiotics are <5%, and even among these persons the organism may be only passing through the gastrointestinal tract following environmental exposure (21,22,23).
In contrast to adults, C. difficile does not have a clear etiologic role for causing symptomatic infection among infants, especially neonates (18,19). Although the exact reason for this is unknown, it may be related to a species-specific innate resistance to infection in early life; while the enterocytes of immature rabbits do not bind toxin A (24), the enterocytes of neonatal pigs do bind the toxin and internalize it (25,26), reflecting the risk of symptomatic infection in one but not the other immature animal population. Regardless, human adults are definitely at risk for symptomatic infection as well as colonization. Humoral immunity is an important determinant of whether an individual with a perturbed microbiome develops infection or only colonization following C. difficile acquisition (17); likewise, immune responsiveness plays an important role in whether a previously infected patient will develop recurrence (27,28).
Supporting the role of humoral immunity in CDI is the finding that patients with asymptomatic colonization and no history of recent symptomatic infection are at decreased rather than increased risk for developing CDI during subsequent hospitalization (29). This paradox, relative to colonization by other multidrug-resistant organisms (MDROs) that generally increase the risk for subsequent infection, reflects the increased levels of antitoxin antibodies that appear in patients with asymptomatic colonization (17). Available data suggest that the incubation period of CDI following transmission/acquisition is <3 days (17,30,31,32), suggesting that persons found asymptomatically colonized have either had preexisting protective levels of antibodies or quickly boosted their antibody levels on the basis of an anamnestic immune response. In a similar vein, recurrent CDI, defined for surveillance purposes as a second episode of symptoms within 8 weeks of initial infection (33), occurs in ~20% of patients with an initial episode of CDI and is associated with a poor antitoxin antibody response to initial infection (27,28). Key evidence confirming the central role of antitoxin antibody levels protecting against recurrence is the recently reported 72% reduction in recurrences observed among patients administered a cocktail of antitoxin A and antitoxin B monoclonal antibodies (34).
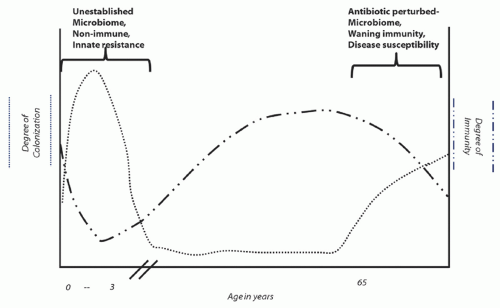 Figure 34.1. Depiction of the prevalence of colonization by C. difficile and evidence of humoral immunity to infection at different stages of life. |
The main symptom of CDI is diarrhea, which may be severe and accompanied by abdominal tenderness, fever, and, in a minority of patients, blood in the stool. Important early indicators of more severe disease that may be more likely to progress to complicated disease include leukocytosis ≥15,000/mm3 and an elevated serum creatinine over baseline (35,36). Leukocytosis to various degrees is quite common, and severe CDI is one of the relatively few infectious causes of white blood cell counts >50,000/mm3 (37,38). While such extreme values can be a poor prognostic sign and ileus can occur in severe disease, CDI is uncommonly an occult cause of leukocytosis in the patient not experiencing diarrhea; leukocytosis alone should not justify
routine C. difficile diagnostic testing in patients with formed stool (16). Pseudomembranous colitis is the classic lesion described in CDI, and can be visualized as white plaques seen on endoscopy or the eruption of cellular debris and pus cells from the mucosa’s deep crypts seen under the microscope. Other histopathologic findings include disruption of the basement membrane with intense infiltration of the mucosa and lamina propria by neutrophils and histiocytes. Not all patients with CDI will have pseudomembranous colitis observable by endoscopy; however, virtually all have diarrhea as their initial and principal manifestation, and this should be the key factor in considering the diagnosis, especially as more sensitive diagnostics are introduced.
routine C. difficile diagnostic testing in patients with formed stool (16). Pseudomembranous colitis is the classic lesion described in CDI, and can be visualized as white plaques seen on endoscopy or the eruption of cellular debris and pus cells from the mucosa’s deep crypts seen under the microscope. Other histopathologic findings include disruption of the basement membrane with intense infiltration of the mucosa and lamina propria by neutrophils and histiocytes. Not all patients with CDI will have pseudomembranous colitis observable by endoscopy; however, virtually all have diarrhea as their initial and principal manifestation, and this should be the key factor in considering the diagnosis, especially as more sensitive diagnostics are introduced.
Complicated disease often involves a progression of severe disease with a thickened edematous colon that may be observed on an abdominal CT scan and may dilate into a condition known as toxic megacolon (39). Meanwhile, the patient may experience a paradoxical slowing of the diarrhea and development of sepsis that presumably reflects translocation of resident bacterial toxins. Although uncommon, perforation can occur and, whether via this mechanism or overwhelming sepsis, death may ensue. Recent carefully performed studies that account for the confounding from patients with CDI often having more severe underlying disease indicate that the attributable mortality of hospital-onset (HO) CDI is between 3% and 5% (40). However, observed mortality in recent outbreaks involving hypervirulent strains has ranged as high as 10% to 15% (41,42).
EPIDEMIOLOGY
The epidemiology of CDI changed dramatically with the emergence of the North American Pulsed Field type 1 (NAP1) strain, also known as the restriction endonuclease analysis (REA) BI strain and PCR ribotype 027 strain (9,41). Although there were previously reported outbreak strains that spread to multiple hospitals, such as the highly clindamycin-resistant REA J-strain responsible for hospital outbreaks in the late 1980s and early 1990s (8,43), none were associated with such sweeping epidemiologic impact as has been noted with NAP1 over the past 12 years (44). It is likely that factors other than strain hypervirulence, such as the aging of Western nation populations and evolving antibiotic usage practices, also have played a role in this changing epidemiology. Although the large outbreak of 2003-4 that affected over a dozen hospitals in Montreal and surrounding environs was the first to make national and international headlines (41), the first known outbreaks caused by NAP1 actually occurred in Pittsburgh in 1999-2000 (45,46) and Atlanta in 2001-2 (47) but were only retrospectively linked to this emerging strain (9). While outbreaks at some centers were clearly associated with increased disease severity (41,45,46), other centers noted no such increase (47). This is most likely due to many confounding factors, such as the median age, underlying severity of illness in the population, and local diagnostic and treatment practices.
Before and early in the emergence of NAP1, only limited surveillance data for CDI from relatively few hospitals in North America and Europe were available (48). However, hospital discharge and death certificate data provide insight into global increases in CDI, first in the United States and Quebec, and later in Europe (49,50,51,52). In the United States, the number of hospitalizations with a discharge diagnosis for CDI more than doubled from 139,000 in 2000 to 347,000 in 2010 (Figure 34.2). Death certificate data during this period reflect an even more startling 4-fold increase from an estimated 3,000 deaths with CDI listed as a cause of death in 1999 to 2000 to 14,000 in 2006 to 2007 (51). While hospital discharge and death certificate data suggest a plateauing of historically high rates in the United States, some countries in Europe have seen a decline in their rates, coincident with a reduction in the proportion of infections caused by NAP1 (53,54).
Estimations of the current U.S. burden of CDI are complicated by the impact of different definitions, settings of onset, and the different diagnostic testing methods discussed below. However, interpolation of existing data suggests the U.S. burden of incident HO-CDI episodes in 2009 to be ~140,000 (52,55). Because HO-CDI represents only 20% to 25% of all CDI (56), it is likely the total U.S. burden for CDI is within range of 500,000 episodes annually. Meanwhile, the attributable costs over 180 days of follow-up for an episode of HO-CDI lie in the range of $5,000 to $7,000, and the attributable excess hospital days are 2.8 days, which, if extrapolated to the above HO-CDI burden estimate, suggests excess U.S. hospital costs of $700 million to nearly $1 billion and nearly 400,000 excess days of hospitalization annually (40,57). However, not only does HO-CDI represent only a fraction of all CDI, there are other outcomes from HO-CDI not reflected in these numbers, such as the proportion of patients who are discharged to a skilled nursing facility instead of to their home—an outcome that is 62% more likely if a patient develops HO-CDI (40).
While the precise molecular factors responsible remain uncertain, there is evidence that, controlling for other factors, NAP1 is responsible for higher rates of disease and more severe disease. In addition to population-based data where the prevalence of NAP1 is associated with higher rates and more severe outcomes in different regions (58), and reductions in these outcomes are temporally associated with decreases in the proportion of CDIs caused by NAP1 (53), results of recent randomized controlled drug treatment trials confirm greater disease severity and likelihood of treatment failure with NAP1 (59). Likewise, recent natural history studies suggest the ratio of symptomatic infections to asymptomatic colonization caused by NAP1 in a population is greater than with other strains (60).
Early on in descriptions of NAP1, it was suggested that increased virulence might be due to increased toxin A and B production resulting from an 18-bp deletion in tcdC, a negative regulator for toxin production within the pathogenicity locus (61). Although an associated upstream frameshift mutation in tcdC has been identified as the responsible genetic change leading to a loss of function (62) and the exact role of tcdC has only grown more controversial (63,64), increased toxin production remains a possible mechanism for hypervirulence (63,65,66,67). The concept of increased sporulation as the main driver for hypervirulence has been refuted (67,68). While binary toxin in NAP1 represents another possible virulence factor that somehow works in concert with toxins A and B (15), polymorphisms in the binding domain of toxin B have also been suggested as a possible virulence factor (69). Moreover, comparative whole genomic sequencing has identified a number of genes that are unique in current NAP1 strains compared to historic NAP1 strains (dating back to the early 1980s) and many more genes that are unique from a standard reference strain (70). Finally, fluoroquinolone resistance in NAP1 appeared coincident to it becoming a cause of outbreaks, and it is likely that the
widespread use of respiratory fluoroquinolones in older adults, a trend that began in the late 1990s, conferred a selective advantage to this strain, leading to its emergence and spread (9).
widespread use of respiratory fluoroquinolones in older adults, a trend that began in the late 1990s, conferred a selective advantage to this strain, leading to its emergence and spread (9).
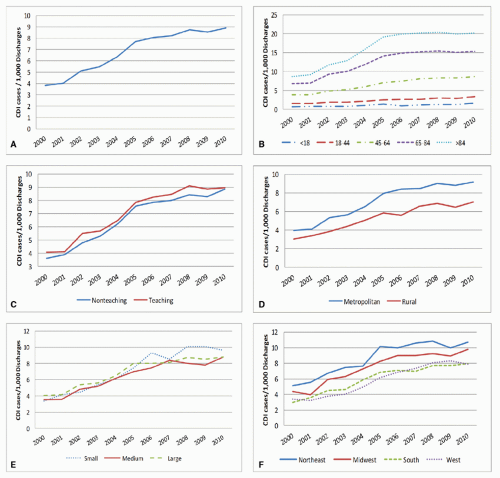 Figure 34.2. Rates of U.S. acute care hospital discharges coded for C. difficile infection (CDI: an ICD9-CM code of 00845 as primary or secondary diagnosis) for years 2000 to 2010, (A) overall and stratified by: (B) patient age category, (C) hospital teaching status, (D) hospital location, (E) hospital size (bed-size criteria vary by region, location, and teaching status), and (F) region. Compiled from the National Inpatient Sample, downloaded at the Healthcare Cost and Utilization Project (available at http://www.hcup-us.ahrq.gov/overview.jsp). |
Around the same time increases were noted in CDI among hospitalized patients the important role of CDI in the community became clearer (71). Approximately 25% to 35% of all CDIs appear to be community-associated (CA)-CDI, meaning that they occur in patients who have had no overnight stay in an inpatient care facility in the previous 12 weeks (56,72). Most remarkable about these patients is that as many as 30% to 40% have not received antibiotics in the previous 12 weeks (73,74), suggesting that factors other than antibiotics may disrupt the microbiome to render a patient susceptible to CDI. In contrast, 90% to 100% of hospitalized patients who develop CDI have received recent antibiotics, and those who have appeared unexposed have been thought by some, at least in the past, to have had occult antibiotic exposures (75,76). The strains responsible for CA-CDI are largely similar to those causing HO-CDI (77,78) except in the Netherlands where another hypervirulent strain, PCR ribotype 078 (NAP 7 and 8), has been found prevalent in rural areas (79). PCR ribotype 078 is of particular interest because it is a common, widespread (e.g., Europe and North
America) cause of sometimes severe CDI in neonatal pigs, raising concern that pathogenic C. difficile could be transmitted via the food supply (79,80,81). However, considering the overall results of recent retail meat studies suggesting little or no contamination and a geographic distribution of human PCR ribotype 078 infections in the Netherlands inconsistent with food distribution patterns (i.e., predominantly rural), available evidence points toward little or no role for foodborne transmission in the epidemiology of human CDIs (79,82,83,84). Nonetheless, there may be a role for amplification in animals of emerging hypervirulent strains such as ribotype 078 with transmission between animals and humans via the environment (79,81,85).
America) cause of sometimes severe CDI in neonatal pigs, raising concern that pathogenic C. difficile could be transmitted via the food supply (79,80,81). However, considering the overall results of recent retail meat studies suggesting little or no contamination and a geographic distribution of human PCR ribotype 078 infections in the Netherlands inconsistent with food distribution patterns (i.e., predominantly rural), available evidence points toward little or no role for foodborne transmission in the epidemiology of human CDIs (79,82,83,84). Nonetheless, there may be a role for amplification in animals of emerging hypervirulent strains such as ribotype 078 with transmission between animals and humans via the environment (79,81,85).
In addition to patients with CA-CDI, who are often younger and have less severe underlying disease than patients with HO-CDI or nursing home-onset CDI (74), episodes of severe CDI and deaths among generally healthy pregnant women have been reported (71,86). However, although there are immunologic reasons to expect that severe CDI could occur in pregnancy and there has been increased antibiotic use associated with prophylaxis for Group B streptococcus, available evidence does not suggest that increases in pregnancy-associated CDI are out of proportion to disease in other populations (87). CDI has also increasingly been recognized among children with onset both inside and outside the hospital. Marked increases in hospital discharge diagnoses for CDI have been noted in the pediatric population as they have in adult patients (88). This includes increases in infants, suggesting that even though there is no good evidence to indicate that C. difficile is etiologic for disease in human infants (89), many clinicians are at least considering the diagnosis and possibly testing and treating for CDI. Above 1 to 2 years of age, C. difficile does appear to cause disease, suggesting that observed increases in children above this age represent increases in true disease similar to that observed in adults (89).
While there is increased recognition of CDI episodes occurring in persons without recent inpatient healthcare exposure (71,72), another way to assess the epidemiology is the proportion of all patients who have had any recent healthcare exposure. Taking this perspective, a recent report from the Centers for Disease Control and Prevention’s (CDC’s) Emerging Infections Program (EIP) population-based surveillance in the United States indicates that 94% of all patients with CDI have had some healthcare exposure within the previous 12 weeks, even if only a recent outpatient exposure (56). Thus, the main focus for preventing CDI across a community should remain focused on healthcare delivery (Figure 34.3). However, because data from the EIP also indicate that 75% of all CDIs related to recent healthcare have their onset outside an acute care hospital, including in nursing homes, rehabilitation facilities, and the community, CDI prevention needs to occur across the continuum of healthcare, suggesting a possibly unique and important role for coordination by state and local public health departments that have relationships with all care settings (56).
All epidemiologic risk factors can be categorized according to the three main prerequisites in pathogenesis of CDI: deficient or waning immunity, microbiome disruption, and transmission leading to new acquisition. Both the incidence of initial CDI and the proportion of patients who experience recurrence are more common with advanced age, most likely related to, among other factors, an age-related loss in ability to mount an anamnestic immune response (27). Recent evidence that a common human genetic polymorphism in the gene encoding interleukin-8 is associated with a deficient humoral immune response to C. difficile toxin A and increased risk for recurrent CDI may constitute the most advanced understanding of a genetic susceptibility to any HAI pathogen (90,91). Similarly, certain cancer patients, patients receiving steroids, and HIV-infected patients are all at increased risk for CDI or severe outcomes independent of antibiotic exposures, most likely via an acquired immune deficiency (75,92,93).
 Figure 34.3. Percentage of C. difficile infection (CDI) cases (N = 10,342), by inpatient or outpatient status at time of stool collection and type/location of exposures in the previous 12 weeks, United States, Emerging Infections Program, 2010. (Adapted from MMWR Morb Mortal Wkly Rep. 2012;61:157-162). |
Now with the dawning of the metagenomics era, it is becoming apparent that the disruption of the lower-intestinal microbiome wrought by antibiotics is both profound and long-lasting (94,95). It has long been recognized that virtually every antibiotic, except perhaps the aminoglycosides, predisposes patients to CDI. It is difficult to precisely rank the CDI risk of different antibiotics because several factors confound the relationship between antibiotic and CDI, including the characteristics of prevalent strains such as hypervirulence or resistance to the antibiotic in question (9), the prevailing “colonization pressure” indicating how likely a patient may newly acquire C. difficile after exposure to the antibiotic (75), and other host factors already mentioned. Given these limitations, it still appears safe to say that broader-spectrum antibiotics, especially those with anaerobic activity and/or against which prevailing strains are highly resistant, all appear to have higher risk than other narrower-spectrum drugs with activity against C. difficile. Thus, advanced-generation cephalosporins, fluoroquinolones, carbapenems, and penicillin-β-lactamase inhibitor combinations appear, somewhat in decreasing order, to be the antibiotics currently in use that place patients at greatest risk per unit of exposure. Overall, antibiotics increase the risk of CDI 7- to 10-fold while a patient is receiving the drug and the month thereafter, and 2- to 3-fold for the 2 months following that (96). In addition, the receipt of multiple antibiotics and cumulative antibiotic exposures over 2 to 3 months increases the risk of CDI more than a single antibiotic administered for a single, short course (97). Similarly, patients who have had a recent episode of CDI and were successfully treated are more likely to have a recurrence if they are again exposed to antibiotics for other reasons (98).
Proton pump inhibitors (PPIs) used in the treatment of peptic ulcer disease and gastric reflux disease, as well as histamine-2 receptor (H2) blockers used for a similar purpose, are risk factors that have only begun to be identified in the past 10 years (99,100). Although there have been several studies that have failed to find an association between these drugs and CDI (73,74), many others have identified this risk, and following several recent meta-analyses all confirming an association, the U.S. Food and Drug Administration (FDA) recently issued a clinician warning about the association (101). Because the spores of C. difficile are quite resistant to the effects of stomach acid and the spore is the predominant infective form, the depletion of stomach acid by PPIs or H2 blockers leading to increased passage of viable organism into the intestine is not a likely mechanism for these drugs to increase CDI risk. Instead, recent data indicate important effect modification between antibiotics and these drugs with larger strength of association for PPIs and H2 blockers and CDI in patients with less intense antibiotic exposures (102). Moreover, PPIs have been demonstrated to impact the lower-intestinal microbiome of dogs (103), suggesting that increased CDI risk from PPIs may result from a perturbation of the lower-intestinal human microbiome in a manner similar to how antibiotics work.
Other epidemiologic risk factors for CDI reflect increasing risk of a patient newly acquiring C. difficile. These include increasing length of stay in an inpatient facility in proximity to source patients who are either colonized or infected with C. difficile (75). In general, it is likely that colonization and infection rates mirror each other across settings such that the incidence of infection in a setting might be an adequate surrogate for “colonization pressure.” Thus, a given patient with a given underlying risk of developing CDI is at higher risk if admitted to a ward or unit with a higher incidence of CDI than a low-incidence setting. Another variation on this risk is the prevalence of active CDI on admission (104). At facilities where a higher proportion of admitted patients have active CDI, whether related to previous care at the same facility or another facility, the risk of other patients acquiring C. difficile from these prevalent patients (and/or the unmeasured number of asymptomatically colonized for which they serve as sentinels) will be greater. The importance of adjusting for this risk in presenting surveillance data will be discussed below. Finally, certain care practices, such as the use of feeding or nasogastric tubes, also increase the risk of CDI (105). While this risk factor may result from increased hand contact by healthcare providers providing increased opportunity for C. difficile transmission, it has been recently suggested that this may reflect the deleterious effects of an elemental diet on the lower-intestinal microbiota (106).
DIAGNOSIS
Currently, there is no test available that can make the diagnosis of CDI but only tests that, combined with a consistent clinical scenario, raise the pretest probability to make a likely diagnosis. While the visualization of pseudomembranes on endoscopy is highly specific for CDI, it is an invasive procedure that is not very sensitive for many patients both because the entire colon may not be visualized (as in flexible sigmoidoscopy) and because many episodes of CDI will not manifest grossly visible pseudomembranes early in their course. Likewise, a biopsy with histopathology that is consistent with pseudomembranous colitis is highly specific but not practical or sensitive.
Historically, the cell cytotoxin neutralization assay (CCNA), a functional assay in which the cytotoxic effect of stool supernatant is demonstrated on a tissue culture cell monolayer and proven due to toxin B via blocking of cytopathic effect by antitoxin B antibodies, was the gold standard for diagnosis (4,5). However, this is a complex test to perform and interpret, requires tissue culture capabilities, and has a 2- to 3-day turnaround. These shortcomings led to the development of rapid and simpler-to-perform enzyme immunoassays (EIAs) for toxin A that are relatively inexpensive and have less than a 1- to 2-hour turnaround. Owing to the emergence of toxin A− B+ positive strains, these early-generation EIAs have been replaced with more sensitive toxin A and B tests. However, even when compared to CCNA, toxin A and B EIAs are insensitive (i.e., ≤80%) although they have fairly high specificity (107,108). Meanwhile, another approach long used in Europe is to culture for the C. difficile organism and, because there are many strains of C. difficile that do not carry toxins A or B and are therefore not pathogenic, a toxin test (i.e., either tissue cytotoxin or EIA) is performed on the isolate (109). While this is much more sensitive in detecting the presence of toxigenic C. difficile, the turnaround time (i.e., 4 to 5 days) makes it less clinically useful, and there are concerns over the detection of asymptomatic carriage if not used in the right population. Nonetheless, toxigenic culture has been proposed as the new gold standard against which to compare the sensitivity of other testing modalities (16).
The past 5 years have witnessed the FDA approval of eight commercial nucleic acid amplification tests (NAATs) (110) each with sensitivities approaching toxigenic culture and turnaround times of a few hours (111). Although these newer technologies are on the whole more expensive than EIAs, with increasing availability of competing platforms, it is anticipated that the cost should become more competitive over time. Meanwhile, use of less-expensive, highly sensitive but nonspecific EIAs for the glutamate dehydrogenase antigen (GDH) has been proposed as a screening test for stools, to be followed by a second confirmatory test (112,113). While a toxin EIA may confirm the diagnosis in many instances, to overcome the poor sensitivity of the toxin EIA, another sensitive method such as a NAAT or CCNA should be used for GDH (+)/EIA (−) samples (i.e., three-step algorithm) (112). Alternatively, a two-step algorithm of GDH with NAAT or CCNA performed on all GDH (+) specimens can be used (113). Including NAAT in such a multistep GDH paradigm may be preferable as the NAAT can be periodically performed in parallel with the GDH to assure the test is performing with desired high sensitivity; there are unconfirmed reports of lower GDH sensitivity that may be strain-specific (114,115). Especially where there is significant delay to final results in a subset of patients (i.e., three-step algorithm), care must be taken in reporting interim results to clinicians in an understandable way.
With the rise in the widespread use of the toxin EIAs, the insensitivity of tests for CDI became well known, leading to reports that repeat testing might bolster sensitivity and widespread adoption of this practice (116). It is now well documented how such repeat testing and other poor test-ordering practices have led to routine testing in populations with very low overall prevalence (i.e., <5%) of true disease. In those patients with a first negative toxin EIA result, the prevalence of true disease is even lower. At such low prevalence, even toxin EIAs with a specificity of 98% can have a positive predictive value of <80%, and repeat
test results quickly approach a positive predictive value as low as 50% (117).
test results quickly approach a positive predictive value as low as 50% (117).
Given such scenarios, it has been recommended that all laboratories migrate toward the use of more sensitive assays while improving the selection of the population in which they are used. It is the opinion of this author that target laboratory positivity rates (i.e., surrogate for prevalence in tested population) should be 7% to 12% if a toxin EIA is still being used, or if, as recommended, a testing paradigm that is more sensitive with a clinically useful turnaround time is used (i.e., two- or three-step GDH algorithm or routine NAAT in all patients), a laboratory positivity rate of 15% to 20% should be targeted. Such positivity rates can be achieved through clinician education regarding the strength (i.e., high negative predictive value) and limitation (i.e., inability of any diagnostic to make the diagnosis of CDI apart from clinical correlation) of the more sensitive approach, education on the appropriate population in which to test (i.e., >3 unformed stools in a 24-hour period) and why not to perform a test for cure (i.e., patients routinely remain colonized following resolution of CDI) (16), and strictly enforced laboratory rejection policies for formed stool and repeat testing within a 5- or 7-day time frame (118).
TREATMENT
General recommendations for the management of patients with CDI include stopping any other antibiotics when possible (whether stopping PPIs makes a difference in clinical outcome is not known), using oral therapy whenever possible, treating for a minimum of 10 days, avoiding antiperistaltics, assuring that anti-C. difficile therapy is reaching the colon in patients with severe disease, and involving surgeons early in the care of patients with complicated disease. Drugs with an FDA-approved indication for CDI include oral vancomycin and fidaxomicin. Meanwhile, metronidazole, although not FDA-approved for CDI, has been a recommended treatment for many years (16). Of these three agents, only metronidazole is orally absorbed and in fact this absorption is quite high in the absence of diarrhea, resulting in low stool concentrations in asymptomatic patients. Because of its absorption, systemic toxicity in the form of peripheral neuropathy can occur if administered for a prolonged period. However, the advantages of metronidazole are its lower cost per course (i.e., owing to its widespread generic availability) and the perhaps mistaken perception that exposure to it vs. oral vancomycin results in less selective pressure for vancomycin-resistant enterococci (VRE) (119).
Oral vancomycin, meanwhile, is not absorbed systemically and has a favorable peak stool concentration relative to minimum inhibitory concentration for C. difficile. In addition to the concern about selecting for VRE, oral vancomycin has always been much more expensive than metronidazole. However, this difference in price may change appreciably with the approval of generic oral vancomycin. Previously, many inpatient settings circumvented the higher price of oral vancomycin by administering generic IV vancomycin via the oral route. However, this practice was less amenable to use in the outpatient setting. Although the equivalence of oral metronidazole and vancomycin was asserted in many previous guidelines, recent trials have shown more favorable outcomes with vancomycin, especially in more severe disease (16,120,121,122). Vancomycin is currently recommended for initial treatment of CDI in patients with early indicators of severe disease, including a WBC >15,000/mm3 and serum creatinine >1 mg/dL over baseline on the date of diagnosis (16).
Fidaxomicin is the most recently FDA-approved therapy for CDI. Like oral vancomycin, fidaxomicin is not systemically absorbed and has favorable activity against C. difficile at concentrations easily achieved in the stool (123). In addition, this drug is thought to cause less disruption to the microbiome (124), and this, along with a demonstrated inhibition of sporulation (125), may be responsible for it being the first approved anti-CDI therapy associated with a lower recurrence rate (126,127
 Epidemiology of Healthcare-Associated Infections
Epidemiologic Methods for Investigating Infections in the Healthcare Setting
Economic Evaluation of Healthcare-Associated Infections and Infection-Control and Antimicrobial-Stewardship Interventions
The Intensive Care Unit, Part B: Antibiotic Resistance and Prevention of CVC-BSIs, Catheter-Associated Urinary Tract Infections, and C. difficile
Nosocomial Tuberculosis
Healthcare-Associated Respiratory Viral Infections
Epidemiology of Healthcare-Associated Infections
Epidemiologic Methods for Investigating Infections in the Healthcare Setting
Economic Evaluation of Healthcare-Associated Infections and Infection-Control and Antimicrobial-Stewardship Interventions
The Intensive Care Unit, Part B: Antibiotic Resistance and Prevention of CVC-BSIs, Catheter-Associated Urinary Tract Infections, and C. difficile
Nosocomial Tuberculosis
Healthcare-Associated Respiratory Viral Infections




Related posts:
Epidemiology of Healthcare-Associated Infections
Epidemiologic Methods for Investigating Infections in the Healthcare Setting
Economic Evaluation of Healthcare-Associated Infections and Infection-Control and Antimicrobial-Stewardship Interventions
The Intensive Care Unit, Part B: Antibiotic Resistance and Prevention of CVC-BSIs, Catheter-Associated Urinary Tract Infections, and C. difficile
Nosocomial Tuberculosis
Healthcare-Associated Respiratory Viral Infections
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
