Diagnostic Implications of Excessive Homozygosity Detected by SNP-Based Microarrays: Consanguinity, Uniparental Disomy, and Recessive Single-Gene Mutations
a Fullerton Genetics Center, Mission Health System, 267 McDowell Street, Asheville, NC 28803, USA
Keywords
• Microarray • Single nucleotide polymorphism • Homozygosity • Autozygosity • Consanguinity • Uniparental disomy
Single nucleotide polymorphism (SNP)-based microarray analysis provides detection of copy number variations (CNVs) as well as genotype information at multiple polymorphic loci throughout the genome. Although the clinical utility of CNV detection is well accepted,1,2 information derived from SNP-based microarray analysis has only more recently been utilized in the constitutional cytogenetics laboratory setting.3,4 In addition to specific genotype data, analysis of SNP allele patterns can provide (1) confirmation of CNV calls, (2) sensitivity for detection of mosaicism, and (3) detection of excessive homozygosity, which is the focus of this review.
Several different SNP-based microarray platforms are currently in clinical use, and this review illustrates similar data obtained from Affymetrix- and Illumina-based microarrays (Affymetrix Inc, Santa Clara, CA, USA; Illumina Inc, San Diego, CA, USA). It is generally not necessary to derive specific genotypes from the microarray data to view the SNP probes as present in a homozygous or heterozygous state. For both platforms, biallelic SNPs are denoted as “A” or “B.” Relative allele distribution is typically provided by either allele difference plots or B-allele frequency plots (Fig. 1).
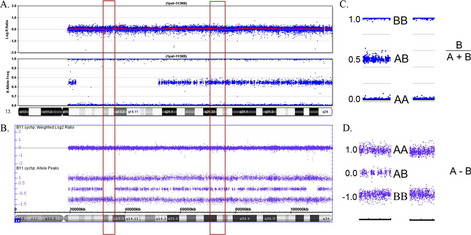
Multiple terms are used to describe regions of homozygosity, and each conveys a slightly different meaning (eg, loss of heterozygosity, absence of heterozygosity, runs of homozygosity). Here we use the term long-contiguous stretch of homozygosity (LCSH) to describe an uninterrupted region of homozygous alleles with genomic copy number state of 2. The term LCSH excludes loss of heterozygosity caused by single copy deletions because these regions exist in a hemizygous state. Minimal thresholds for LCSH calls are generally set around 0.5 to 1 Mb in population genetic analyses5–7 and more conservatively at 3 to 10 Mb in clinical analyses,4(Kearney H, Kearney J, and Conlin L, personal communication).
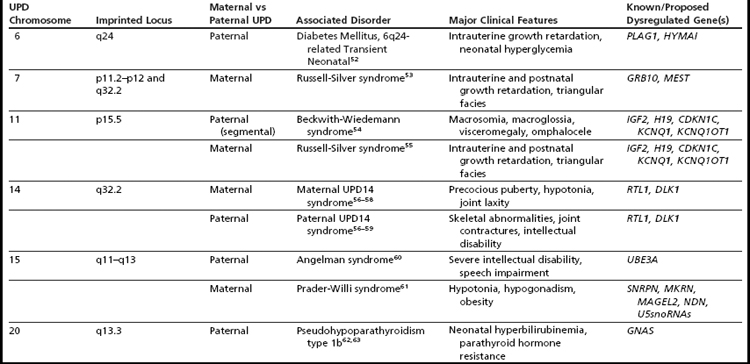
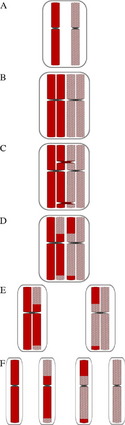
Detection of excessive homozygosity, in and of itself, is not diagnostic of any underlying condition and may be clinically benign. The first step in assessing the clinical relevance of observed LCSH is to distinguish between excessive homozygosity found in multiple regions throughout the genome versus LCSH restricted to a single chromosome. When multiple LCSH regions are found throughout the genome, the findings are generally assumed to represent regions identical by descent (IBD), with the associated concerns for recessive disorders mapping to the homozygous intervals. When the genomic homozygosity is sufficiently excessive, the finding may trigger a suspicion for parental consanguinity or incest (Table 1; Fig. 2).8 One or more regions of LCSH found only on a single chromosome can be a hallmark of uniparental disomy (UPD), either whole-chromosome UPD or segmental UPD,3,4,9 and may warrant further clinical investigation, particularly when involving chromosomes associated with imprinted gene disorders (Table 2). Regardless of mechanism or size, all LCSH segments have the potential to harbor homozygous recessive mutations.
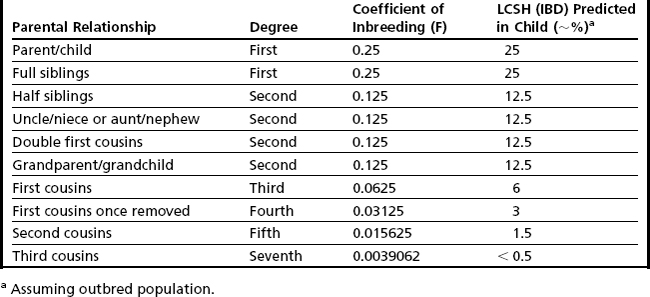
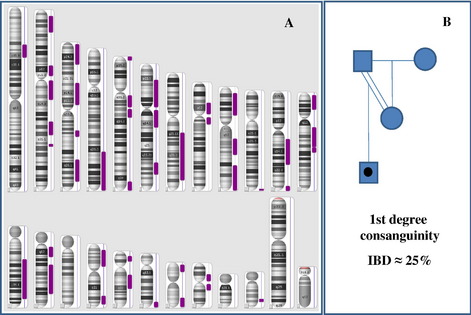
Fig. 2 LCSH patterns seen in consanguinity. (A) Male patient with LCSH on multiple chromosomes (Affymetrix Genome-Wide Human SNP 6.0 array data visualized in Affymetrix Chromosome Analysis Suite [ChAS] software). Note that the sex chromosomes are excluded in LCSH modeling. Calculations of total percentage IBD with different minimal LCSH thresholds in this case result in the following estimations: LCSH threshold ≥ 0.5 Mb: 868,341 kb (30% IBD); ≥ 1 Mb: 868,341 kb (30%); ≥ 3 Mb: 866,089 kb (30%); ≥ 5 Mb: 858,676 kb (30%); ≥ 10 Mb: 797,445 kb (28%); ≥ 15 Mb: 700,140 kb (24%), and ≥ 20 Mb: 577,279 kb (20%). These data and those of additional cases suggest that minimal LCSH thresholds between 0.5 and 10 Mb have negligible impact on estimation of percentage of IBD. These data are consistent with first degree consanguinity, as illustrated in panel B (and Table 1). (B) Pedigree illustrating first-degree consanguinity and predicted percentage IBD. The mother and father of the proband share 50% of their genome (because they are first-degree relatives). There is a one-half probability that the proband will inherit an identical genomic region from both of his parents, making the total estimate of the proband’s percentage of IBD in this scenario roughly 25%. Note that other first-degree parental relationships (eg, full siblings) would also yield an estimated 25% IBD in the proband.
Consanguinity
When LCSH is found distributed throughout the genome, this observation is presumed to represent homozygosity caused by inheritance of genomic regions of IBD. When the parents of a proband share a recent common ancestor, their union is defined as consanguineous. The closer the parental relationship, the greater the proportion of shared alleles and, therefore, the greater the risk of the child (proband) inheriting 2 copies of a deleterious gene mutation from his parents.10–12 Clinical laboratories that perform SNP-based microarray analysis will encounter many cases of presumed parental consanguinity, occasionally with suspicion of abuse/incest because of the degree of consanguinity estimated (Fig. 2).8
Although this has no immediate clinical utility and represents a pursuit largely of academic or social/ethical/legal interest, an estimate of the total proportion of the LCSH in the genome can be used as a rough assessment of degree of parental relationship (see Table 1). A simple method to grossly estimate parental relationship is to add all homozygous regions greater than a defined threshold (eg, 3 Mb), excluding the sex chromosomes (because males are always hemizygous, and X chromosomes in females are often observed with increased LCSH because of more limited recombination). The total autosomal LCSH can then be divided by total autosomal length (2,867,733 kb for hg18) to estimate the percentage of IBD. This estimation can then be correlated with the predicted percentage of IBD for various degrees of relationship (see Table 1). It should be noted that this crude calculation is likely to represent an underestimate of the actual homozygous proportion given that (1) only LCSH long enough to be detected by with the array platform/applied threshold will be included and (2) the denominator includes regions of the genome that may not be covered on the microarray (eg, acrocentric short arm and centromeric regions). Although in theory, the background level of population-specific homozygosity in any genome may artificially inflate this observation, in practice, this does not complicate the calculated percentage to any measurable degree, particularly if the threshold used to calculate total LCSH is greater than 1 Mb.6,7
Estimating percentage of IBD can be illustrative and suggestive of degree of parental relationship, but if this inference is made at all, it should be accompanied by an appropriate measure of uncertainty. Most importantly, this estimate cannot be taken as evidence of a specific parental relationship. Additionally, this inference assumes a standard 50% distribution of parental alleles in each meiotic division, and unpredictable recombination and chromosome segregation patterns may result in significant deviation from this distribution.7 Furthermore, consanguinity is very common practice in some populations13,14; therefore, it is likely that individuals from these populations share not just a single recent ancestor but also multiple common ancestors (eg, total genomic homozygosity near or exceeding that seen with first-degree consanguinity, yet the parents have a fairly distant relationship). The laboratory generally has limited or no information regarding the family/social/ethnic situation of the proband; therefore, inferences regarding suspected abuse involving a parent are generally poorly supported; any communications regarding suspicion of abuse should follow appropriate professional guidelines and take place under advisement of one’s institutional legal/ethics consult. Currently, there are no professional guidelines for whether concerns for abuse/incest should be revealed after SNP-array analysis and, if so, under what circumstances, although guidance from the American College of Medical Genetics is forthcoming. Regardless of whether consanguinity is suspected, estimated, or even revealed, simply informing the referring physician of increased suspicion for recessive disorders has clinical utility (see Autozygosity Mapping section).
Uniparental Disomy
Generally, when isolated LCSH (involving only a single chromosome) is detected, particularly when longer than 10 Mb,4 uniparental disomy is considered a likely mechanism. UPD is defined as the inheritance of both homologues from a single parent.15,16 Many excellent reviews have been devoted to UPD,17–24 and there are informative Web-based resources available as well (Lier lab site, Jena University Hospital25; Morrison lab site, University of Otago26; Robinson lab site, University of British Columbia27; Jirtle lab site, Duke University28). This review will, therefore, not cover the history, exhaustive mechanisms, or specific clinical features of UPD syndromes (see Table 2 for summary), but will instead focus on the laboratory detection of UPD through SNP-based microarray analysis and appreciation of associated data complexities. Historically, UPD was only suspected when accompanied by a hallmark cytogenetic finding (mosaic trisomy, marker chromosome, or other structural rearrangement, such as a Robertsonian translocation), by clinical manifestation of a disorder of imprinting,29 or finding homozygosity for a recessive allele with only a single carrier parent.16 Through the use of SNP-based microarrays, clinical laboratories may now have serendipitous detection of unanticipated UPD events by recognition of hallmark patterns of homozygosity.4 To appreciate the expected patterns of homozygosity encountered with UPD-involved chromosomes, it is useful to review the various mechanisms known to generate UPD. It is of fundamental importance to first appreciate the basic concepts of meiosis and meiotic recombination, which are summarized in Fig. 3.
There are 2 primary mechanisms by which UPD involving a whole chromosome may be generated: (1) trisomy rescue, the most frequently observed mechanism and (2) monosomy rescue.23 Gamete complementation has also been proposed as a UPD mechanism,15 but this is thought to be a very rare event. Additionally, segmental UPD (involving only part of a chromosome) may also be generated through somatic events.17 Given that the majority of these mechanisms generate UPD with full or partial homozygosity of parental markers (Figs. 4–6), SNP-based microarrays are very useful for detecting LCSH patterns that may be predictive (but not diagnostic) of UPD.3,4,9,24 Uniparental isodisomy refers to inheritance of 2 identical chromosomes from a single parent, whereas uniparental heterodisomy refers to inheritance of 2 homologous chromosomes from the same parent. Because of meiotic recombination, even UPD events involving an entire chromosome are usually not purely isodisomic or heterodisomic, but instead often have a mixture of both types of segments.

Related posts:
 Cytogenetics
Cytogenetics
 Structural Rearrangements: Detection and Elucidation of Mechanisms Using Cytogenomic Technologies
Structural Rearrangements: Detection and Elucidation of Mechanisms Using Cytogenomic Technologies
 Evolving Picture of Microdeletion/Microduplication Syndromes in the Age of Microarray Analysis: Variable Expressivity and Genomic Complexity
Evolving Picture of Microdeletion/Microduplication Syndromes in the Age of Microarray Analysis: Variable Expressivity and Genomic Complexity
 In Situ Hybridization
In Situ Hybridization
 Syndromes
Syndromes
 Myeloid Leukemia: Conventional Cytogenetics, FISH, and Moleculocentric Methodologies
Myeloid Leukemia: Conventional Cytogenetics, FISH, and Moleculocentric Methodologies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


