Immunomodulatory Drugs and Proteasome Inhibitors
The therapeutic options for the treatment of multiple myeloma (MM) have expanded over the past decade with the introduction of novel biologically targeted agents, which in turn have resulted in significantly improved outcomes (1). The use of the immunomodulatory drug thalidomide in the 1990s and subsequently its analogue lenalidomide and more recently pomalidomide has been a major advance in the field. The proteasome inhibitor, bortezomib, was FDA approved in 2003, and the next-generation proteasome inhibitors are now undergoing evaluation in clinical trials. Antibodies targeting membrane-bound receptors are another promising class of agents.
IMMUNOMODULATORY DRUGS (IMiDs)
Thalidomide was originally developed as an antihistamine, but produced significant sedation and was marketed as a hypnotic. It alleviated symptoms of morning sickness due to pregnancy. However, in 1963 it was withdrawn when its use was associated with stunted limb growth (dysmelia) in children born of women exposed to the drug during pregnancy. Three decades later it was found to improve signs and symptoms of erythema nodosum leprosum. This approval reawakened interest in its antiangiogenic and immunomodulatory effects and led to its successful trial for the treatment of patients with refractory MM (2).
Lenalidomide was developed as a thalidomide analogue that more effectively inhibited TNF-α. Pomalidomide is the newest IMiD.
 STRUCTURE
STRUCTURE
Thalidomide is piperidinyl isoindole ([±]-α-[N-phthalimido]) glutarimide. It is a neutral racemic compound derived from glutamic acid, and is structurally related to the analeptic drug bemegride (α-ethyl-α-methyl-glutarimide, C18H13NO2), and to a sedative and antiepileptic drug, glutethimide (β-ethyl-β-phenyl-glutarimide, C15H23NO4). It has two ring systems: a left-sided phthalimide, and a right-sided glutarimide with an asymmetric carbon atom at position 3′ of the glutarimide ring. The drug consists of equimolar amounts of (+)-R- and (–)-(S)-enantiomers. Thalidomide is sparingly soluble in water (<0.1 g/l) and spontaneously hydrolyses in solution at pH 6.0 or higher to produce at least 12 different products. The structure of thalidomide was modified through the addition of an amino group at the 4 position of the phthaloyl ring, to generate pomalidomide and with the further removal of a carbonyl on the ring to form lenalidomide (Figure 6-1).
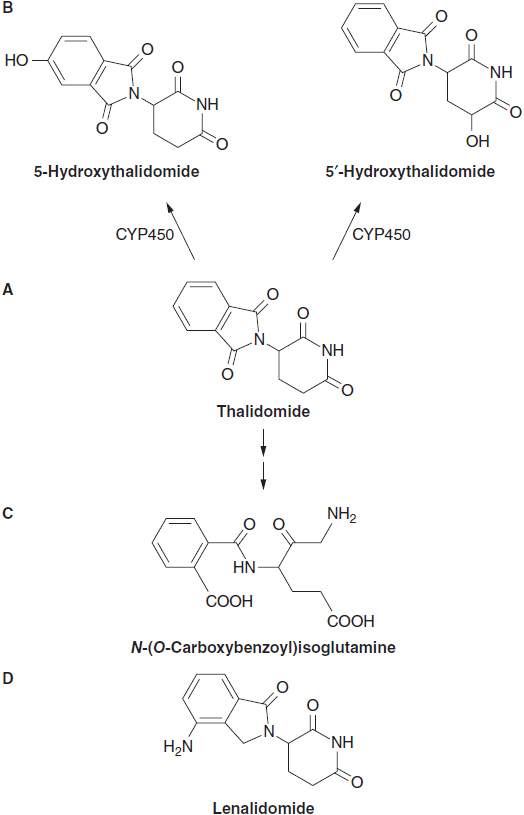
FIGURE 6-1 The structure of thalidomide, hydroxylated metabolites with their hydrolytic products and analogs lenalidomide and pomalidomide. (A) Thalidomide, (B) its p450 hydroxylation product, (C) a spontaneous hydrolytic breakdown product, and (D) its analog, lenalidomide.
 MECHANISM OF ACTION
MECHANISM OF ACTION
IMiDs are hypothesized to act through multiple mechanisms including:
1. A direct antiproliferative/proapoptotic effect probably mediated by downregulation of TNFα, a tumor stimulating and immunomodulatory cytokine, and a suppressive effect on nuclear factor kappa B (NF-κB), a transcription factor that promotes a protective response to cell injury.
2. An indirect antitumor effect mediated by downregulation of tumor cell adhesion molecules (I-CAM 1) and stimulatory cytokines such as IL-6.
3. Inhibition of secretion of angiogenic cytokines such as basic FGF and VEGF.
Its teratogenic activity is attributed to its binding to and inactivating a ubiquitin ligase, cereblon, the suppression of which aborts limb development in zebrafish (3). The presence of cereblon is required for the antimyeloma activity of the IMiDs (4).
 METABOLISM AND CLINICAL PHARMACOLOGY
METABOLISM AND CLINICAL PHARMACOLOGY
Thalidomide is slowly absorbed from the gastrointestinal tract of human subjects due to its poor aqueous solubility, with peak levels in plasma at 3–6 h after ingestion. Absorption at doses at or below 200 mg is variable but becomes linear as doses increase to 800–1200 mg. Thalidomide is loosely bound to serum albumin and to α1-acidic glycoprotein and is widely distributed throughout the body. It is mainly broken down through nonenzymatic hydrolytic cleavage, but about 20% of the drug is also metabolized by hepatic CYP2C19 to form hydroxylated metabolites (Figure 6-1).
The extent of hydrolysis has been estimated to be 80% by 24 h. Hydrolysis cleaves the two-imide bonds of both enantiomers, opening the glutarimide and phthalimide rings, and yielding a series of 12 products, 11 of which are chiral (5). These products are further broken down to yield numerous optically active compounds.
Thalidomide disappears from plasma with an apparent half-life of 4.7 h for both the (S)- and (R)-enantiomers, which are rapidly interconvertible in vivo. Thalidomide and its metabolites are excreted in the urine, while the nonabsorbed portion of the drug is excreted unchanged in feces. Clearance of the parent compound is primarily metabolic and non-renal. Studies of multiple oral doses in healthy subjects showed that thalidomide capsules 200 mg/day over 18–21 days did not produce accumulation and the AUCs on days 1 and 21 were equivalent. It is not known if accumulation occurs with higher daily dosages, although pharmacokinetic simulations of 400 and 800 mg once daily suggest that this would not occur (6).
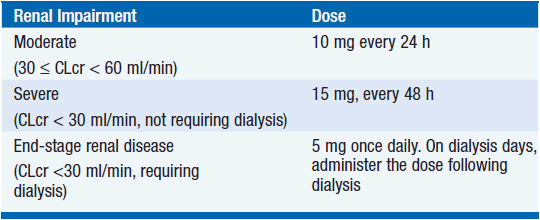
Lenalidomide is absorbed rapidly after oral intake, reaches maximum plasma concentration after 1–1.5 h, and disappears from plasma with a halflife of 3.1–4.2 h. Approximately two-thirds of the drug are excreted intact in the urine; its renal clearance exceeds glomerular filtration. The remainder of the drug is excreted unchanged in feces, with little evidence for metabolism. Dose adjustments are required in the presence of renal insufficiency (Table 6-1) (7).
TABLE 6-1 RECOMMENDED DOSE ADJUSTMENTS OF LENALIDOMIDE FOR PATIENTS WITH MM WITH IMPAIRED RENAL FUNCTION
 TOXICITY
TOXICITY
Sedation and constipation are the most important side effects of thalidomide and become dose limiting at daily doses of 400 mg or greater. Other side effects include dizziness, mood changes, headaches, skin rash, and, rarely, neutropenia. Peripheral neurotoxicity, primarily peripheral sensory changes, is reported in up to one-third of patients, usually at doses of 400 mg or greater. Hematologic toxicity, primarily a decrease in the neutrophil and platelet counts, is modest and seen only at doses of 400 mg or greater. As a single agent, thalidomide causes a 5% incidence of deep vein thromboses, but both lenalidomide and thalidomide in combination with dexamethasone cause major thrombotic events in up to 15% of patients, especially in these receiving 400 mg doses of thalidomide or greater (8).
Lenalidomide does not cause significant sleepiness, or constipation, and neuropathy is infrequent. However, it causes neutropenia and thrombocytopenia in approximately 20% of patients. It enhances myelosuppression caused by concomitant chemotherapy, and predisposes to venous thrombosis and embolism when used with dexamethasone.
Prophylactic anticoagulation should be considered in all patients receiving an IMiD particularly in combination with glucocorticoids. The International Myeloma Working Group panel recommends the use of aspirin in patients with one risk factor for VTE. Low-molecular-weight heparin (equivalent to enoxaparin 40 mg/day) is recommended for patients with two or more risk factors for VTE (8).
In clinical trials, second malignancies are more common in patients receiving lenalidomide maintenance compared to patients receiving placebo. McCarthy et al. reported 8 new hematologic cancers and 10 solid-tumor cancers (excluding nonmelanoma skin cancers) among the 231 patients in the lenalidomide group (3.5% and 4.3%, respectively) compared to 1 new hematologic and 5 solid-tumor cancers among the 229 patients in the placebo group (0.4% and 2.2%, respectively) (9). Factors predisposing to second malignancies are being evaluated (10).
The major toxicity of pomalidomide is neutropenia. Grade 3 or greater neutropenia has been seen in 26%–66% of patients, depending on the dose and intensity of prior treatment (11–13). Thromboembolic complications occur with a frequency similar to lenalidomide. Neuropathy is infrequent, with some worsening reported in heavily pretreated patients, most of whom had neuropathy at baseline. Noninfectious acute lung injury is a rare but serious complication that responds well to glucocorticoids (14).
CLINICAL EFFECTIVENESS
Thalidomide is approved for the treatment of newly diagnosed MM patients. Because of thalidomide’s known teratogenicity, the FDA restricts marketing of thalidomide in the United States via the System for Thalidomide Education and Prescribing Safety (S.T.E.P.S.®) program.
Lenalidomide is approved by the U.S. Food and Drug Administration for use in combination with dexamethasone in patients with MM who have received one prior therapy. It is also approved for patients with 5q-myelodysplastic syndrome. Lenalidomide is available under a special restricted distribution program, called RevAssistSM.
Pomalidomide is approved for treatment of relapsed or refractory myeloma.
There is emerging data supporting the use of lenalidomide as maintenance treatment after autologous stem cell transplantation (9, 15, 16).
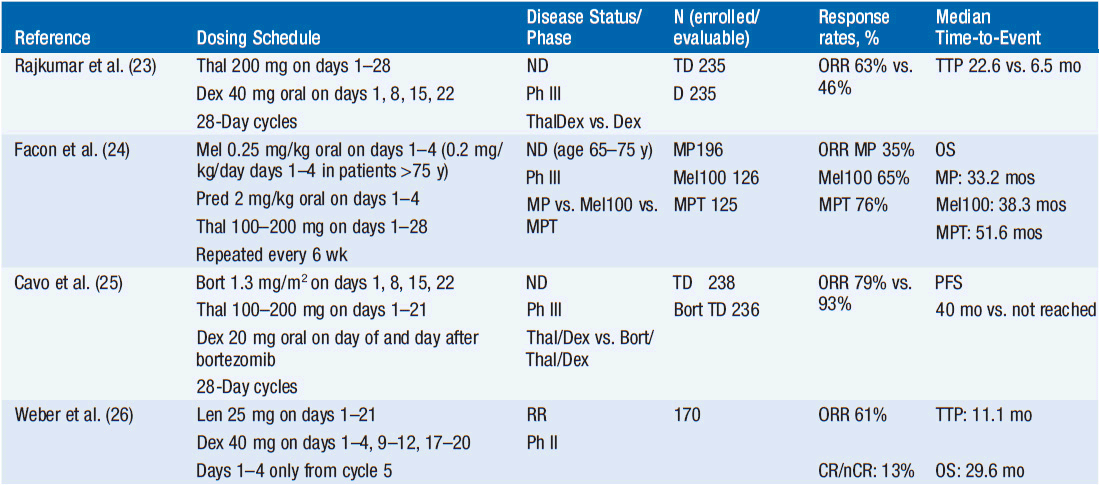
Selected clinical trials with the above drugs are summarized in Table 6-2.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


