Immunology of Hematopoietic Stem Cell Transplantation
Srinivas Viswanathan, Yi-Bin Chen
Hematopoietic stem cell transplantation (HSCT) has wide applications in the treatment of hematologic malignancies, congenital and acquired disorders of hematopoiesis, and autoimmune disease. Transplanted hematopoietic progenitors can reconstitute the full spectrum of hematopoietic cells in a recipient host, and can also confer immunologic antitumor activity. HSCT is therefore employed for both cellular replacement and for cancer immunotherapy. This chapter reviews the basic immunology of HSCT.
MAJOR CELLULAR TYPES
Following HSCT, donor hematopoietic progenitor cells migrate to the recipient bone marrow and differentiate to generate all cell types of the erythroid, myeloid, and lymphoid lineages. The following hematopoietic cell types are thought to play key physiologic roles:
• CD34+ cells: The population of cells containing the hematopoietic progenitors capable of repopulating the recipient marrow. HSCT grafts are usually quantified based on the CD34+ population. In certain selective cellular depletion protocols, donor hematopoietic cells can be purified for CD34+ cells prior to transplantation. Doses of more than or equal to 2 × 106/kg (recipient weight) CD34-expressing progenitor cells are typically required in autologous or adult allogeneic adult donor products. Requirements are approximately one log lower for umbilical cord products.
• B-cells: Lymphocytes whose chief role is to produce antibodies against specific antigens to mediate humoral immunity. The role of B cells in chronic graft-versus-host disease (cGVHD) has been recognized.
• CD8+ T cells: Cytotoxic T cells that recognize antigenic peptides presented on major histocompatibility (MHC) Class I proteins. In the presence of either a costimulatory signal or stimulatory cytokines (secreted by CD4+ Th and other cells), CD8+ T cells undergo clonal expansion and lyse their targets via the release of perforin and granzyme.
• CD4+ helper T (Th) cells: Helper T cells that can develop into either Th1 or Th2 effector T cells. Th1 cells support the cellular immune response by stimulating killing by macrophages and CD8+ T cells. Th2 cells support the antibody response by stimulating B-cell proliferation and antibody production. A third subset of Th cells (Th17 cells) has been identified although its role in HSCT is yet to be defined.
• Natural killer (NK) cells: Cytotoxic lymphocytes central to the innate immune response. NK cells kill cells that have lost cell-surface expression of self MHC I in a perforin- and granzyme-dependent manner. NK cells are also key effectors in humoral immunity, mediating antibody-dependent cellular cytotoxicity (ADCC).
• Regulatory (Treg) cells: A subset of CD4+ T cells that functions in a regulatory capacity to modulate the immune response and suppress autoimmunity. Ongoing clinical trials are exploring different ways to expand Treg cells to control GVHD.
• Dendritic cells: Antigen-presenting cells (APCs) that express high levels of both MHC I and MHC II and efficiently present antigens to T cells. Dendritic cells stimulate naïve T cells within lymphoid tissues. Dendritic cells within the thymus eliminate T cells that are selective for self-antigens through the process of negative selection.
GRAFT-VERSUS-HOST DISEASE
Graft-versus-host disease (GVHD) results from an immunologic response against antigenic disparities between donor hematopoietic cells and host tissues. GVHD comprises a significant proportion of morbidity and mortality associated with HSCT. Historically, GVHD had been classified into acute (traditionally occurring within 100 days of transplant) and chronic (traditionally occurring after 100 days of transplant) phases. However, recent consensus definitions have transitioned to a classification based solely on clinical manifestations, rather than the timing of symptoms after HSCT. There is also an acute-chronic GVHD overlap syndrome which has manifestations of both classic acute and chronic disease (Table 37-1).
Significant acute GVHD (aGVHD) complicates up to half of all HLA-matched stem cell transplants and a higher proportion of mismatched transplants. The most commonly involved sites of aGVHD are the skin, liver, and gastrointestinal tract. Clinical features include a maculopapular rash, liver function test abnormalities (traditionally, elevations in direct bilirubin and alkaline phosphatase), and high-volume diarrhea accompanied by abdominal cramping. Histologic features differ by organ, and it is worth noting that the overall clinical picture, and not pathologic findings, is the gold standard for diagnosis. Skin involvement by aGVHD is characterized histologically by dermal and epidermal lymphocytic infiltration. Liver aGVHD is characterized by lymphocytic infiltration of small bile ducts, leading to bile duct damage and degeneration, and intestinal aGVHD is characterized by crypt cell necrosis, increased apoptosis, and a loss of intestinal epithelium (1, 2).
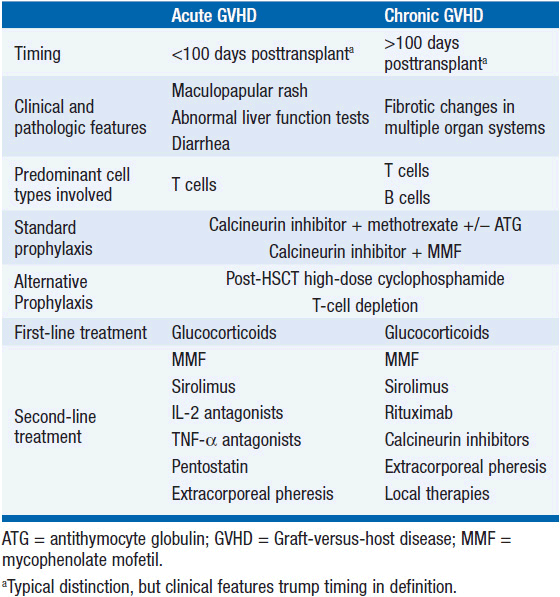
The pathogenesis of aGVHD is thought to be driven by donor T-cell-mediated damage of host cells as well as by local and systemic release of inflammatory cytokines. This is thought to occur via a multistep process: (1) Damage to host tissues by the conditioning regimen (classically, high doses of chemotherapy and radiation) stimulates the release of proinflammatory cytokines including IL-1, IL-6, TNF-α, and IFN-γ (2) increased MHC molecule expression on host APCs including dendritic cells in response to cytokines; (3) presentation of alloantigens to donor T cells by activated host APCs triggers T-cell activation, leading to IL-2 and IFN-γ production; (4) CD4+ T cells stimulate expansion of the donor CD8+ T cells that mediate the cytotoxic effects of GVHD; and (5) direct damage by the cytotoxic actions of activated donor CD8+ T-cells and inflammatory cytokines result in the clinical manifestations of acute GVHD (Figure 37-1).
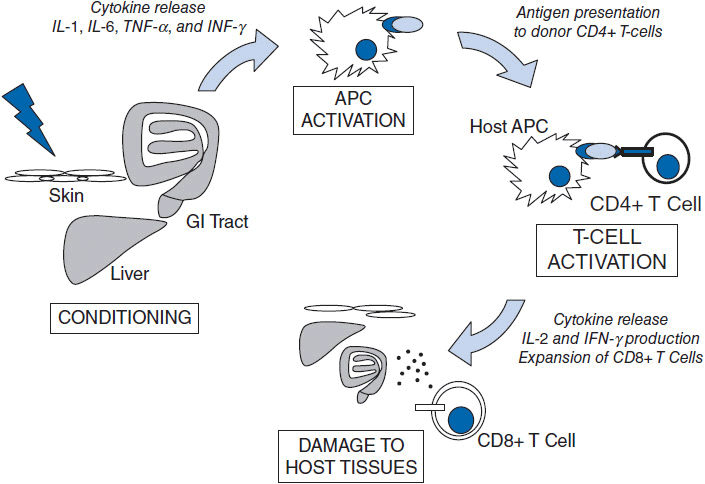
Figure 37-1 Schematic of the steps leading to GVHD. In the afferent phase, damage to host tissues by conditioning regimen leads to inflammatory cytokine production and upregulation of MHC on host APCs. Subsequently, recipient and donor APCs activate donor-derived T cells. Cytotoxic (CD8) T cells then cause damage to host tissues.
Every allogeneic HSCT requires some form of prophylaxis against GVHD. This can be accomplished through (1) pharmacologic methods (most commonly) or (2) ex vivo T-cell depletion methods. The accepted international standard for pharmacologic prophylaxis against aGVHD involves a calcineurin inhibitor (either cyclosporine or tacrolimus) in combination with several low doses of post-HSCT methotrexate. Calcineurin inhibitors are generally continued at therapeutic levels for several months with gradual tapering to discontinuation if there are no signs of GVHD.
The initial treatment for patients who develop clinical aGVHD despite appropriate prophylaxis involves high-dose systemic glucocorticoids, usually at doses of 1–2 mg/kg/day of prednisone or its equivalent. Glucocorticoids provide durable remission in only 50% of patients, and those who fail initial therapy have high mortality rates (3, 4). There is no standard second-line therapy and a number of agents have been tried, including mycophenolate mofetil, sirolimus, IL-2 antagonists, TNF-α antagonists, pentostatin, and extracorporeal pheresis.
Chronic GVHD (cGVHD) develops in a subset of patients with acute GVHD, and also arises in the absence of any preceding acute GVHD. The most commonly involved sites are the eyes, skin, respiratory tract, esophagus, and liver. Clinically, chronic GVHD is pleomorphic and heterogeneous, with many features resembling classic autoimmune diseases such as lupus, Sjogren’s syndrome, vitiligo, and scleroderma. In contrast to the inflammatory changes that are typically found in aGVHD biopsies, pathologic hallmarks of cGVHD include a dense fibrosis of involved tissues with occasional infiltration of mononuclear cells (5).
Both donor B cells and donor T cells appear to play important roles in the pathophysiology of chronic GVHD. T-cells have been implicated in cGVHD in multiple mouse models, as well as by the fundamental observation that cGVHD incidence is significantly less in patients who undergo transplants from T-cell depleted grafts. The role of B cells has emerged recently from studies showing antibodies specific for proteins coded from the Y chromosome in female donor—male recipient transplants (6). In addition, other studies have also demonstrated that B-cell activating factor (BAFF), a pro-B-cell growth factor, is present at high levels in patients with active chronic GVHD (7).
Many of the same agents used for treatment of aGVHD are also used for the treatment of cGVHD, including systemic glucocorticoids, calcineurin inhibitors, mycophenolate mofetil, sirolimus, and extracorporeal pheresis. Given the recent data implicating the role of B cells in cGVHD, the anti-CD20 antibody rituximab has also been employed with encouraging results (8). Chronic GVHD has emerged as the most important determinant of quality of life in long-term survivors of HSCT (9), and, unfortunately, many trials that have shown some success in reducing acute GVHD have not had a significant effect on preventing chronic GVHD. Therefore, one of the primary focuses of future research in HSCT involves better prevention and treatment of chronic GVHD.
GRAFT-VERSUS-LEUKEMIA
The existence of an immunologic graft-versus-leukemia (GVL) effect has been supported by several observations: (1) with comparable conditioning regimens, allogeneic transplants result in a lower relapse rate than autologous or syngeneic transplants; (2) multiple series have observed that patients who develop acute or chronic GVHD have a lower incidence of disease relapse than those who do not (10); and (3) donor leukocyte infusions (DLI) alone were able to achieve remission for many patients with relapsed chronic myeloid leukemia (CML) after HSCT (11). Although antitumor effects of HSCT have been most widely studied in the setting of leukemia, regression of other hematologic malignancies has also been clearly described following HSCT, leading to the use of the broader term graft-versus-malignancy (GVM). The recognition of the GVM effect has led to the increasing popularity of protocols employing nonmyeloablative and reduced-intensity conditioning regimens. The efficacy of these protocols is based mostly on the antitumor effects of GVM, with toxicity spared due to the lower intensity of the conditioning regimens.
The extent or potency of GVM appears to vary based upon the underlying disease, disease status, and, undoubtedly, unknown essential interactions between the donor and the recipient. Both donor-derived T cells and NK cells appear to have key roles in GVM. In theory, the T-cell response is driven by the presentation of host or tumor antigens to donor T cells, which leads to a clonal expansion of CD8+ cytotoxic T cells specific for the antigen. NK cells also appear to be potent mediators of GVL (12, 13), but differ from T cells in that they can kill tumor cells without the prerequisites of activation and clonal expansion. NK cells express inhibitory receptors known as killer cell immunoglobulin-like receptors (KIRs) which recognize specific inhibitory MHC Class I allele groups (KIR ligands). NK-cell alloreactivity occurs when recipient cells do not express an MHC-I ligand which can engage KIR on the surface of donor NK cells. Overall, it remains unclear if the immunologic mechanisms driving GVM differ from those responsible for GVHD, and current methods are unable to reliably separate the two (14). Ongoing research is focusing on methods to better cultivate GVM without inducing significant GVHD, using approaches such as preemptive post-HSCT treatment with immunomodulatory agents and vaccination protocols against specific tumor antigens. A summary of these approaches is shown in Figure 37-2.
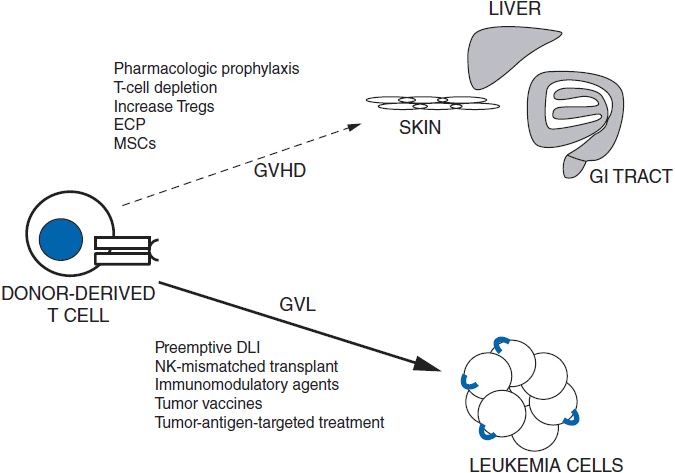
FIGURE 37-2 The promise of HSCT lies in augmenting GVL (thick arrow) while minimizing GVHD (dotted arrow). Strategies for minimizing GVHD include pharmacologic agents, T-cell depletion, increasing number of Treg cells, extracorporeal pheresis (ECP), and mesenchymal stem cell (MSC) infusion. Strategies for augmenting GVL include donor-lymphocyte infusion (DLI), increasing number of Treg cells, conducting NK-cell (KIR) mismatched transplants, tumor vaccines, and therapies targeted and tumor-specific antigens.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


