Introduction
Glucose is an obligate fuel for the brain under physiological conditions. The brain can neither synthesize glucose nor store more than approximately a 20-minute supply of glycogen; therefore brain survival requires a continuous supply of glucose. The brain can use alternative fuels from the circulation, provided their concentrations rise high enough to enter the brain in sufficient amounts; for example, elevated ketones during prolonged fasting or lactate during vigorous exercise and in untreated type 1 glycogen storage disease (GSD). Blood-to-brain glucose transport mediated by glucose transporter 1 (GLUT-1) is a direct function of the arterial plasma glucose concentration, and physiologic mechanisms normally maintain plasma glucose at levels that ensure adequate glucose delivery to the brain. At physiological (70–100 mg/dL ) plasma glucose concentrations, the rate of blood-to-brain glucose transport exceeds the rate of brain glucose metabolism. At plasma glucose concentrations lower than 54 mg/dL, however, the cerebral metabolic rate of glucose decreases; and at even lower plasma glucose concentrations, functional brain failure occurs, and profound and prolonged hypoglycemia causes permanent brain injury and eventually brain death.
Beyond the newborn period and early infancy, hypoglycemia is uncommon and is usually caused by an acquired disorder of the endocrine system, prolonged fasting in susceptible individuals (e.g., an intercurrent gastrointestinal illness), congenital abnormalities, such as hyperinsulinism (HI) or inborn errors of metabolism, or accidental and rarely deliberate exposure to medication or toxins. As the interval between feedings increases in the growing infant, physiologic endocrine and metabolic processes normally ensure the maintenance of normoglycemia; however, hypoglycemia can first manifest later in infancy or early childhood when there are mild congenital defects of these systems. In contrast, hypoglycemia presenting in older children and adults is typically caused by an acquired disorder.
This chapter describes an approach to diagnosis based on identifying the specific cause of failure to maintain normal glucose homeostasis. Key diagnostic information is often best derived from blood and urine specimens (referred to as critical samples ) obtained at the time of hypoglycemia and immediately before reversing hypoglycemia.
Physiologic development of glucose metabolism during infancy and childhood
Glucose Production and Utilization
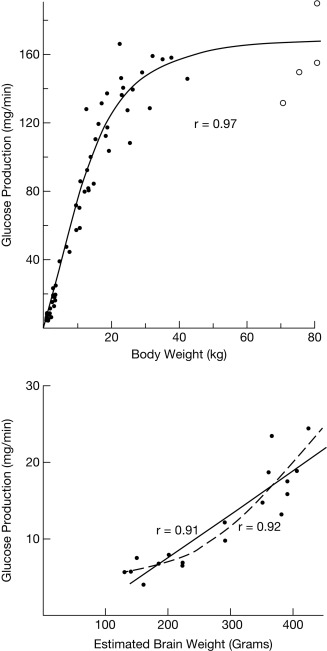
Rates of glucose flux into and out of the circulation are normally tightly regulated and systemic glucose balance is maintained, while ensuring a continuous supply of glucose to the brain. After a typical carbohydrate-containing meal, increased insulin secretion (together with inhibition of counterregulatory hormone secretion) leads to rapid disposal of ingested glucose, either for immediate energy needs or storage via deposition of glycogen and conversion to fat, resulting in restoration of plasma glucose concentrations to basal levels within 2 to 3 hours. Bier et al. showed that in children, ranging from premature infants to age 6 years, mean glucose production rates are 5 to 8 mg/kg/min; thereafter, glucose production as a function of body weight decreases toward the mean adult value of 2.3 mg/kg/min. Furthermore, the rate of glucose production is linearly correlated with estimated brain weight at all ages. Because the brain accounts for the bulk of daily glucose utilization, it is not surprising that adult glucose production rates are reached by mid-childhood (body weight ~ 30 kg), when the child’s brain weighs about 90% of the adult brain ( Fig. 23.1 ).
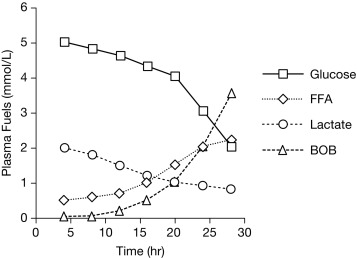
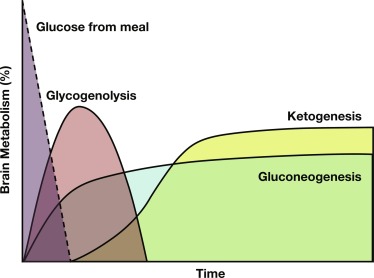
During fasting, the brain initially uses glucose almost exclusively provided from a combination of glycogenolysis and gluconeogenesis in the liver. As liver glycogen stores diminish, adipose tissue lipolysis is activated to increase availability of free fatty acids (FFA) as fuel for peripheral tissues, such as muscle, and for ketogenesis in the liver, which makes ketones available as a brain fuel and partly replaces glucose utilization. Note that the time to reach the fasting hyperketonemia stage is markedly shorter in younger infants and children than in adults, because of their larger ratio of brain relative to muscle mass. As shown in Fig. 23.2 , the change in sources of fuel for the brain is reflected in the changes in plasma concentrations of the major metabolic fuels. The substantially smaller muscle mass of infants and young children relative to brain mass limits their ability to tolerate prolonged fasting. Because brain growth is nearly complete by 10 to 12 years of age, plasma glucose can be maintained above 70 mg/dL for progressively longer fasting durations in adolescents and adults. Nonetheless, most full-term infants, from about 1 week to 1 year of age, are able to tolerate fasting for 15 to 18 hours before plasma glucose decreases below 70 mg/dL, and by 1 year of age, most normal children are able to fast for up to 24 hours. By 5 years of age, a fast of up to 36 hours may be tolerated, and most adults can maintain fasting plasma glucose above 70 mg/dL for 48 to 72 hours ( Fig. 23.3 ). When hypoglycemia is induced by fasting for a shorter duration than would be expected for the child’s age, the possibility of an underlying hypoglycemia disorder should be considered.
Adaptation to Longer Feeding Intervals
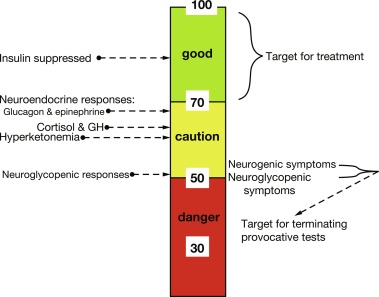
With feeding, plasma insulin concentrations in normal-weight children rise from basal levels of 3 to 10 μU/mL to peak levels of 20 to 50 μU/mL, which serves to stimulate hepatic glycogen synthesis, inhibit hepatic glycogenolysis and gluconeogenesis, and enhance peripheral (muscle and adipose tissue) glucose uptake and utilization ( Table 23.1 ). Simultaneously, triglyceride synthesis is activated and lipolysis and ketogenesis are suppressed. In the postabsorptive state, a characteristic sequence of physiological responses are involved in preventing or correcting hypoglycemia. The earliest response to decreasing plasma glucose concentrations is a decrease in insulin secretion, as plasma glucose declines within the physiological range (80–85 mg/dL) followed by increased secretion of glucose counterregulatory hormones (glucagon, epinephrine, cortisol, and growth hormone [GH]), when plasma glucose falls just below the postabsorptive physiological range (65–70 mg/dL), and sympathetic neural activation occurs at 55 mg/dL ( Fig. 23.4 ). The combined effects of suppressed insulin secretion, increased levels of counterregulatory hormones, and sympathetic neural activation mobilize stored fuels and reduce glucose utilization, thereby preventing hypoglycemia. With insulin levels suppressed, elevated glucagon secretion and sympathetic neural activation trigger glycogenolysis, the first phase in the metabolic defense against hypoglycemia. In infants, liver glycogen stores may provide glucose for up to 4 hours (see Fig. 23.3 ). As the child grows, hepatic glycogen content relative to brain glucose utilization is greater and may be able to provide glucose for up to 8 hours of fasting. Isolated glucagon deficiency is extremely rare and, with the exception of children on beta-blocker drugs, deficiency of sympathetic nervous activity is equally unusual. Therefore hypoglycemia occurring within 2 to 4 hours of a feed suggests either a primary disorder of glycogenolysis or excess (dysregulated) insulin secretion. As glycogen stores become depleted, maintenance of plasma glucose levels relies on gluconeogenesis and reduced tissue glucose utilization (resulting from increased oxidation of FFA and ketones) (see Fig. 23.3 ). The main gluconeogenic precursors are amino acids, especially alanine and glutamine, from skeletal muscle, and glycerol from adipose tissue lipolysis ( Fig. 23.5 ). FFA become the body’s major fuel source, as the duration of fasting becomes more prolonged. Mitochondrial fatty acid oxidation in the liver produces ketone bodies (acetoacetate and β-hydroxybutyrate [BOHB]), which are used for energy production by the brain and skeletal muscle, including cardiac muscle (see Fig. 23.2 ). Oxidation of FFA and ketone utilization decreases peripheral glucose utilization, which helps to ensure an adequate supply of glucose to the brain and tissues that can only use glucose as fuel (e.g., red blood cells and renal medullae), while preventing excessive breakdown of muscle protein. Lipolysis of triglycerides in adipose tissue is triggered by low levels of insulin together with increased secretion of the counterregulatory hormones (especially GH ) and sympathetic neural activation. In infants, elevation of plasma ketone concentrations begins within 12 to 18 hours of fasting; whereas, in older children, substantial ketonemia may not appear until 18 to 24 hours of fasting. Cortisol, secreted in response to stress, accelerates gluconeogenesis and lipolysis and decreases glucose utilization.
| Insulin | Glucagon | Epinephrine a | Cortisol | Growth hormone | |
|---|---|---|---|---|---|
| Glucose uptake | + | – | – | – | |
| Glycogenolysis | – | + | + | b | b |
| Gluconeogenesis | – | + | + | + | + |
| Lipolysis | – | + | + | + | |
| Ketogenesis | – | + | + | + |
a includes sympathetic nervous system activation
b cortisol stimulates glycogen synthesis, and both cortisol and growth hormone exert permissive effects on the gluconeogenic and glycogenolytic effects of glucagon and epinephrine.
Defects in gluconeogenesis (e.g., fructose-1,6-bisphosphatase deficiency; see Fig. 23.5 ) usually become manifest only after glycogen stores have been depleted; accordingly, hypoglycemia typically does not occur in the recently fed state. Disorders of fatty acid oxidation also typically manifest after more prolonged fasting. In the first few months of life, the interval between feedings in the on-demand breastfed infant gradually increase from about 2 to 3 hours to 4 hours or more, and eventually to 8 to 12 hours as nighttime feedings are omitted. Therefore disorders of gluconeogenesis and fatty acid oxidation seldom present as hypoglycemia in the newborn period when feeding is frequent; rather, they present later in infancy as the interval between feeds becomes more prolonged. They may, however, present immediately after birth before lactation is established. Congenital or acquired deficiencies of cortisol and GH may also cause hypoglycemia either in the newborn period, if congenital and severe, or later in infancy when longer periods of fasting typically occur. Combined deficiency of cortisol (adrenocorticotropic hormone [ACTH]) and GH in hypopituitarism may cause earlier onset and more severe hypoglycemia than occurs with isolated deficiencies of either GH or cortisol.
Symptoms, signs, and effects of hypoglycemia
Control of the counterregulatory responses is at least partially local: a low glucose concentration and a decrease in intraislet β-cell insulin secretion stimulate glucagon secretion. Low plasma glucose concentrations are sensed in the hepatic portal vein, as well as in the gut, carotid body, and oral cavity (and are transmitted to the brain) and also are directly sensed in the hindbrain and hypothalamus, which initiates an increase in sympathoadrenal activity.
The trigger for counterregulatory responses is the plasma glucose level itself. The rate at which glucose decreases and the absolute level of insulin have little effect. The symptoms of hypoglycemia, which reflect the brain’s response to glucose deprivation, have been extensively studied and well characterized in adults ( Box 23.1 ). Autonomic (neurogenic) symptoms primarily arise from perception of the physiological changes caused by sympathetic (not adrenomedullary) neural activation and include both adrenergic (tremor, palpitations, anxiety/nervousness) and cholinergic responses (sweating, hunger, paresthesias). Awareness of hypoglycemia chiefly depends on perception of the central and peripheral effects of these neurogenic (in contrast to neuroglycopenic) responses to hypoglycemia. Neurogenic symptoms are perceived at a plasma glucose of less than 54 mg/dL, the threshold at which brain glucose utilization becomes limiting. In older children and adults, the search for food or assistance is a crucially important defense against more severe hypoglycemia. Plasma glucose below 50 mg/dL causes neuroglycopenic manifestations (the result of an insufficient supply of glucose to maintain brain energy), such as impaired cognition, loss of motor coordination, confusion, coma, and seizures. Hypothermia often occurs with prolonged hypoglycemia in older children and adults and is thought to be the result of a neurogenic mechanism. It is noteworthy that young children (6–11 years old) with type 1 diabetes have poor ability to detect low blood glucose levels.
Neurogenic symptoms caused by activation of autonomic nervous system
- •
Sweating
- •
Shakiness, trembling
- •
Tachycardia
- •
Anxiety, nervousness
- •
Weakness
- •
Hunger
- •
Nausea, vomiting
- •
Pallor
- •
Hypothermia
Neuroglycopenic symptoms caused by decreased cerebral glucose use
- •
Headache
- •
Visual disturbances
- •
Lethargy, lassitude
- •
Restlessness, irritability
- •
Difficulty with speech and thinking, inability to concentrate
- •
Mental confusion
- •
Somnolence, stupor, prolonged sleep
- •
Loss of consciousness, coma
- •
Hypothermia
- •
Twitching, convulsions, “epilepsy”
- •
Bizarre neurologic signs
- •
Motor disturbances
- •
Sensory disturbances
- •
Loss of intellectual ability
- •
Personality changes
- •
Bizarre behavior
- •
Outburst of temper
- •
Psychologic disintegration
- •
Manic behavior
- •
Depression
- •
Psychoses
- •
Permanent mental or neurologic damage
Activation thresholds for the neuroendocrine responses have been best established for healthy young adults and vary only slightly by sex, age, exercise, sleep, and nutritional status. There is no evidence that the definition of hypoglycemia should be lower in infants and young children. Indeed, both epinephrine and GH are released at a higher plasma glucose level in children than in adults, suggesting that the threshold for neuroendocrine responses to decreasing plasma glucose levels in children is not lower and may actually be higher than in adults. At comparable plasma glucose thresholds, epinephrine levels are also markedly increased in children as compared with adults. Drugs alter the neuroendocrine responses to hypoglycemia, for example, beta-blockers, dampen the response, whereas caffeine amplifies the response. Prior exposure to hypoglycemia and hyperglycemia cause clinically important alterations of neuroendocrine response thresholds. Even a single episode of moderately severe hypoglycemia can blunt or lower the activation threshold for 24 hours or more; after prolonged or recurrent hypoglycemia, autonomic responses may be sufficiently attenuated that neuroglycopenic effects may be the sole clinical manifestation of severe hypoglycemia, a condition referred to as hypoglycemia-associated autonomic failure ( HAAF ). HAAF has been demonstrated in adults and children with type 1 diabetes, in nondiabetic adults with an insulinoma, and in infants with recurrent hypoglycemia caused by congenital HI. Conversely, chronic hyperglycemia is associated with higher glucose thresholds for counterregulatory responses.
The effects on cognition, behavior, and level of consciousness are typically completely reversed when the glucose level is raised, although subtle neuropsychological impairment may be measurable days later. Severe and prolonged neuroglycopenia, however, causes brain injury and neuronal death. In experiments with primates, plasma glucose below 20 mg/dL for 5 to 6 hours reliably produces severe injury. In both infants and adults, magnetic resonance imaging (MRI) shows characteristic changes caused by hypoglycemia-induced brain injury. Pathologic changes are observed particularly in cortical tissue and white matter; whereas, the cerebellum and brainstem tend to be spared. Permanent cognitive impairment is common both in children and adults after recurrent, severe hypoglycemia.
Definition of hypoglycemia
Clinical hypoglycemia is defined as a plasma glucose concentration low enough to elicit defensive neuroendocrine responses, which cause symptoms and/or signs, or impair brain function. Because the signs and symptoms are nonspecific, hypoglycemia may be difficult to recognize, and a single low plasma glucose concentration may be an artifact. For example, when blood is drawn but plasma not immediately separated from the cellular elements, glycolysis in red cells causes glucose levels to decline at a rate of 6 mg/dL/h. This is a common cause of artifactually low glucose levels reported in metabolic panels measured at commercial laboratories. Point-of-care glucose meters, originally designed for diabetes management, are useful for screening purposes, but their accuracy is limited to approximately ± 10 to 15 mg/dL in the hypoglycemia range. Whole blood glucose levels are approximately 15% lower than plasma concentrations, and it is preferable consistently to refer to plasma glucose concentrations. Before establishing a diagnosis of hypoglycemia and undertaking a diagnostic evaluation, it is essential to confirm a low plasma glucose concentration using a clinical laboratory method. For these reasons, guidelines emphasize the value of Whipple’s triad for confirming hypoglycemia : (1) symptoms and/or signs consistent with hypoglycemia, (2) a documented low plasma glucose concentration, and (3) relief of symptoms and signs when plasma glucose is restored to normal. Because infants and young children cannot dependably recognize or communicate their symptoms, recognition of hypoglycemia may require confirmation by repeated measurements of plasma glucose concentration and formal provocative testing.
It is important to appreciate that hypoglycemia cannot be defined as a single specific plasma glucose concentration because thresholds for specific brain responses to hypoglycemia occur across a range of plasma glucose concentrations, and these thresholds can be altered by the presence of alternative fuels, such as ketones, and also by recent antecedent hypoglycemia. It is also not possible to identify a single plasma glucose value that causes brain injury, and the extent of injury is influenced by other factors, such as the duration and degree of hypoglycemia, availability of alternative fuels, and potential artifacts and technical factors that lead to inaccuracies in glucose measurements.
Despite the frequency with which hypoglycemia is not accompanied by obvious symptoms in infants, evidence of injury to the brain from prolonged or recurrent hypoglycemia suggests the same clinical thresholds and treatment goals are applicable irrespective of age beyond the immediate (48–72 hours) newborn period. Two different hypoglycemia thresholds are recommended for use in clinical practice. To determine the etiology of hypoglycemia and terminate a diagnostic provocative fasting test, obtain critical samples when plasma glucose is less than 55 mg/dL. For therapeutic purposes, however, the lower limit of the plasma glucose range should be 70 mg/dL (see Fig. 23.4 ). This goal is important to avoid periods of low glucose that may blunt the neuroendocrine and symptomatic responses to hypoglycemia and lead to greater susceptibility to subsequent more severe episodes of hypoglycemia.
As noted earlier, the effects of specific plasma glucose levels on the central nervous system may vary among patients, especially after experiencing previous episodes of hypoglycemia. Because there is no exact correspondence between the risk of harm (including brain injury) and either severity of symptoms or specific plasma glucose levels, hypoglycemia should always be treated urgently.
Fasting systems approach to the diagnosis of hypoglycemia
To provide appropriate treatment for a hypoglycemia disorder, it is essential to establish a specific diagnosis of the etiology. Because most hypoglycemia disorders, especially in infants and children, involve a disturbance of fasting adaptation, the best approach to diagnosis is a closely monitored fasting test, which both reproduces the hypoglycemia and surveys the integrity of the major metabolic and endocrine systems shown in Figs. 23.3 and 23.5 and in Table 23.1 . These “fasting systems” include the four metabolic systems (hepatic glycogenolysis, hepatic gluconeogenesis, adipose tissue lipolysis, and hepatic ketogenesis) and their regulation by the endocrine system. The endocrine system includes: (1) insulin, which suppresses activity of all four metabolic systems; and (2) the four counterregulatory hormones that oppose insulin action in specific tissues: (a) glucagon acts acutely on liver to stimulate glycogenolysis and gluconeogenesis and facilitate ketogenesis; (b) adrenergic nervous system activation (reflected by increases in plasma epinephrine levels) acts acutely on adipose tissue and liver to stimulate lipolysis, ketogenesis, glycogenolysis, and gluconeogenesis; (c) cortisol has longer term effects required for maintaining liver glycogen stores and promoting gluconeogenesis; and (d) GH, which has longer-term effects on the capacity for adipose tissue lipolysis (see Table 23.1 ).
The operation of these metabolic and endocrine systems is reflected by changes in major circulating metabolic fuels during normal fasting, as shown in Fig. 23.2 . As liver glycogen reserves become depleted, plasma glucose falls toward hypoglycemic levels, plasma lactate declines by 25% to 50% (a marker of enhanced gluconeogenesis), plasma FFA rise 4- to 6-fold (a marker of lipolysis), and plasma ketones rise 20- to 40-fold (a marker of ketogenesis, usually measured as BOHB the major ketone). Measurement of these fuels at the time of hypoglycemia (the critical samples) provides crucial information about which component of the fasting systems is responsible for causing hypoglycemia ( Fig. 23.6 ). For example, abnormal elevation of plasma lactate suggests a defect in gluconeogenesis; abnormal suppression of BOHB, with a normal or excessive rise in FFA, suggests a defect in fatty acid oxidation; abnormal suppression of both FFA and BOHB suggests excessive insulin action; and a normal fall in plasma lactate accompanied by a rapid rise in FFA and ketone concentrations occurring after a relatively brief period of fasting (i.e., ketotic hypoglycemia) suggests a defect in glycogenolysis. Cortisol or GH deficiency usually manifest with ketotic hypoglycemia; however, in the neonatal period, combined pituitary hormone deficiencies, especially, may present as hypoketotic hypoglycemia, mimicking HI. Deficiency of sympathetic nervous activity (e.g., secondary to beta-blocker drugs) may present with hypoketotic hypoglycemia because of impaired lipolysis. Deficiency of glucagon as a primary disease has not been described, whereas secondary deficiency is common in patients with recurrent hypoglycemia, such as congenital HI or type 1 diabetes (HAAF).
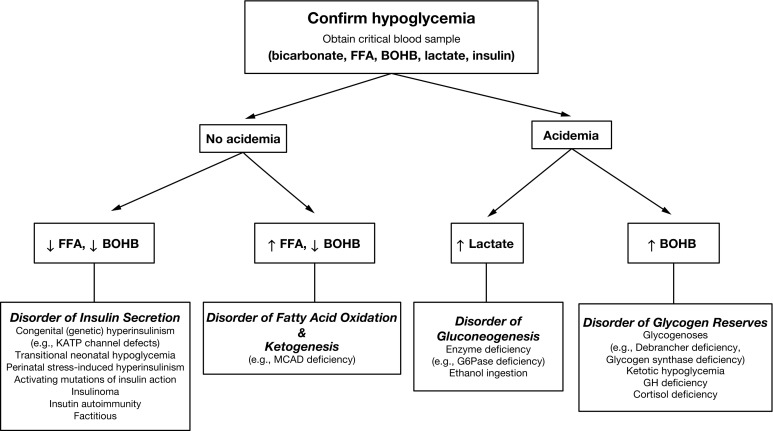
Additional tests may be done separately to further assess for specific disorders or can be conveniently added at the end of the fasting test by using extra blood obtained with the critical sample. Serum insulin levels are often not elevated enough for diagnosis of HI disorders; therefore insulin levels, within the normal range, do not exclude the possibility of HI; plasma C-peptide levels may more accurately reflect pancreatic insulin secretion because insulin delivered by the portal vein is variably extracted by the liver, whereas C-peptide passes through the liver without hepatic extraction. HI can be conveniently confirmed with a glucagon stimulation test: in normal children, there is little or no response at the time of hypoglycemia, because liver glycogen stores have been depleted; whereas in HI, there is an abnormally large glycemic response caused by inhibition of glycogenolysis. Plasma levels of cortisol or GH sometimes increase to values sufficiently high during hypoglycemia to exclude deficiency of either or both; however, before making a diagnosis of cortisol or GH deficiency, low values must be confirmed by additional specific provocative tests of these hormones. Plasma free and total carnitine levels, together with the profile of plasma acylcarnitines, are useful for the diagnosis of most, but not all, of the fatty acid oxidation defects.
Other tests may be considered if specific disorders are suspected. Plasma ammonia concentrations are elevated 3- to 5-fold in patients with the HI hyperammonemia syndrome caused by activating mutations of GLUD1 . Plasma C-peptide suppression in the face of elevated insulin levels may be helpful in demonstrating exogenous insulin administration. Plasma insulin-like growth factor binding protein-1 (IGFBP-1) concentration usually rises 5- to 10-fold as insulin levels decline during fasting, so that low IGFB-1 values support a diagnosis of HI. Specimens to test for drugs or toxins may be considered in cases of suspected ingestion of oral hypoglycemic drugs, plant toxins (unripe ackee-ackee fruit, litchi fruit), or surreptitious insulin administration (factitious hypoglycemia or Munchausen by proxy).
Detailed methods for performing fasting tests and other tests useful for diagnosis of hypoglycemia are provided at the end of this chapter.
Major causes of hypoglycemia in the toddler and child
Hyperinsulinism
HI is the most common and the most severe form of hypoglycemia in neonates and young infants, and is also one of the most common causes of hypoglycemia in toddlers and older children. Hypoglycemia caused by HI is particularly dangerous to the brain, because glucose deficiency cannot be compensated for by increased production of ketones to support brain metabolism. Although many neonates with severe congenital HI present in the first week of life, milder genetic forms of HI continue to be an important cause of hypoglycemia throughout infancy. Older children and adolescents with HI may have acquired insulin-secreting pancreatic adenomas.
Congenital HI may present beyond the first months of life as feeding intervals lengthen and overnight feedings are omitted. Such infants may have early morning lethargy or seizures and may have a history of previous seizures and unexplained developmental delay, or of hypoglycemia in the newborn period that was not fully evaluated or treated. In dominant forms of HI, there may be a history of hypoglycemic symptoms in a parent or other relatives; however, carriers may often not recognize symptoms. As outlined in Chapter 7 , all of the genetic causes of HI may initially present in the neonatal period.
The diagnosis of HI is suggested when hypoglycemia is not accompanied by hyperketonemia and can be confirmed when injection of glucagon causes a large increase (> 30–40 mg/dL relative to baseline) in plasma glucose concentration. HI may be suggested by increased rates of glucose utilization (glucose infusion rate [GIR] > 8 mg/kg/min), but this is less consistently present in older children than in neonates. A critical specimen obtained during hypoglycemia, which demonstrates increased levels of insulin or C-peptide can be conclusive; however, it is often not possible to demonstrate clearly elevated insulin levels. Table 23.2 shows the diagnostic criteria for hyperinsulinemic hypoglycemia. In cases of surreptitious insulin administration, standard insulin immunoassays may not detect exogenously injected insulin analogs, such as lispro, aspart, glargine, etc. In suspected cases, specific assays must be requested.
|
Adenosine Triphosphate Sensitive Potassium Channel Hyperinsulinism
Mutations in the adenosine triphosphate-sensitive potassium (KATP) channel genes are the most commonly identified genetic defects in patients with congenital HI. The ABCC8 and KCNJ11 genes on chromosome 11p encode the two subunits, SUR1 and Kir6.2, of the KATP channel in the plasma membrane of the beta-cell. Potassium efflux through this channel hyperpolarizes the beta-cell plasma membrane and is a key negative regulator of insulin secretion; genetic defects which impair KATP channel activity and preclude channel opening cause persistent depolarization of the plasma membrane and uncontrolled insulin secretion. As described in Chapter 7 , there are three distinct forms of KATP-HI:
- •
Biallelic inheritance of two recessive KATP mutations causes diffuse HI with severe neonatal onset that is unresponsive to diazoxide.
- •
Recessive KATP channel mutations also can cause focal congenital HI by a two-hit mechanism, involving a paternally transmitted recessive KATP mutation combined with an embryonic somatic mutation in the corresponding maternal gene, resulting in effective paternal 11p uniparental isodisomy for the region containing both KATP and the Beckwith-Wiedemann imprinted genes. The resulting focal area of islet cell adenomatosis contains beta cells with defective KATP channels, resulting in HI.
- •
Dominantly inherited KATP channel mutations act in dominant-negative fashion to produce diffuse HI with KATP channels that have varying degrees of impaired channel function. Most dominant KATP mutations are diazoxide responsive, but diazoxide-unresponsive mutations do occur.
The diffuse and focal forms of KATP-HI account for more than 90% of diazoxide-unresponsive cases of congenital HI in early infancy, and are similar in their clinical manifestations. Both may require surgery to maintain safe glucose levels.
Beyond the neonatal period, dominant KATP mutations are more common and are often diazoxide-responsive.
Diazoxide (5–15 mg/kg/day) is the first-line drug for treatment of HI, but is often ineffective for KATP-HI, because it acts as a KATP channel opener. Octreotide may be given by subcutaneous injection at 5 to 15 mcg/kg/day. Longer acting somatostatin analogs, such as lanreotide, have recently been introduced, with some success in older infants. Calcium-channel blockers are not effective. If medical management cannot maintain the plasma glucose concentration greater than 70 mg/dL, with a normal feeding schedule, surgery may be necessary. Surgical treatment for focal KATP-HI can be curative, whereas for diffuse disease even a 95% to 99% pancreatectomy may not cure the hypoglycemia, and insulin-dependent diabetes is a frequent consequence. 18F-fluorodopa positron-emission tomography ( 18 F-L-DOPA PET) scans have been accurate in preoperative localization of a focal lesion.
Glutamate Dehydrogenase Hyperinsulinism
HI can be caused by dominant activating mutations in GLUD1 on 10q, which encodes glutamate dehydrogenase (GDH). This disorder is also known as the HI hyperammonemia syndrome , because the increased GDH enzymatic activity in kidney tubules results in increased renal ammoniagenesis. Hypoglycemia in GDH-HI is usually milder than in KATP-HI, and less likely to present in the newborn period. As is common with dominant genetic disorders, up to 80% of cases with GDH-HI have a de novo mutation. The hyperammonemia in GDH-HI appears to be asymptomatic. However, affected children frequently have brain manifestations, possibly because of increased GDH activity in neurons and altered levels of glutamate or other neurotransmitters: these include absence seizures (generalized epilepsy), behavior disorders, and mild to moderate developmental delay.
In GDH-HI, activating mutations of GDH in the beta cell amplify leucine-triggered production of ATP, leading to closure of KATP channels and insulin release, independent of glucose levels. In affected children, ingestion of high-protein feedings can induce hypoglycemia (leucine-sensitive or protein-sensitive hypoglycemia). In the kidney, the same mutations increase oxidation of glutamate to alpha-ketoglutarate with the release of ammonia.
The diagnosis of GDH-HI is similar to that of other forms of HI; however, persistently elevated plasma ammonia levels (typically 80–120 μmol/L) are specific for this disorder. Because the hyperammonemia is of renal origin, plasma ammonia levels are unrelated to either insulin secretion or plasma glucose levels and are also not related to protein feeding. Thus elevated ammonia levels can be readily demonstrated in casual (random) specimens of plasma. Most patients respond well to diazoxide and do not need surgery.
Glucokinase Hyperinsulinism
Glucokinase (encoded by GCK ) is the enzyme that serves as the glucose sensor in the beta cells of the pancreas. Gain-of-function glucokinase mutations result in a lower glucose threshold for insulin secretion, leading to persistent hypoglycemia (in contrast, GCK loss-of-function mutations produce a higher glucose threshold for insulin release, causing a common form of mild monogenic diabetes (MODY2). At least 15 different dominantly expressed GCK-HI mutations have been reported. Alterations in the enzyme kinetics of individual mutations predict widely different degrees of lowering of glucose threshold, but do not correlate with clinical severity and course. In some cases, GCK-HI mutations present with neonatal hypoglycemia, severe enough to require pancreatectomy to control hypoglycemia; however, some mutation carriers have escaped recognition even into adulthood.
GCK-HI may be especially challenging to diagnose, because fasting plasma glucose levels tend to stabilize at a glucose threshold between 50 and 65 mg/dL. During fasting, if plasma glucose remains at this threshold for an extended period, levels of FFA and BOHB can rise, suggesting a form of ketotic hypoglycemia rather than HI. Plasma ammonia levels are normal in GCK-HI patients and they do not have protein-sensitive hypoglycemia. The response to diazoxide has been only partial and transient in most patients with GCK-HI and some have required pancreatectomy. One report has described good results using a ketogenic diet to prevent symptomatic hypoglycemia; the long-acting somatostatin analog, lanreotide, might be useful in older patients, but experience is limited.
Hexokinase 1 Hyperinsulinism
Hexokinase 1 ( HK1 ) is closely related to glucokinase and carries out the first step in glucose oxidation by phosphorylating glucose to glucose-6-phosphate in most tissues of the body, except for beta cells. In contrast to glucokinase, which has a low affinity for glucose (Km ~ 5 mM) that sets the threshold for glucose stimulation of insulin secretion at around 4 to 4.5 mM (70–80 mg/dL), HK1 has a very high affinity for glucose (Km ~ 1 mM), and its expression in beta cells is therefore normally disallowed. Two reports of congenital HI possibly because of beta-cell expression of HK1 have recently been published. The first report described a large, dominant pedigree with diazoxide-responsive congenital HI, in which linkage analysis identified a shared haplotype on chromosome 10 that included 3 noncoding mutations in the HK1 locus. This family was in one of the original descriptions of congenital HI by McQuarrie in 1953. It was suggested that one or more of these mutations caused HK1 to be expressed in beta cells and to reduce the glucose threshold for insulin secretion to around 55 to 60 mg/dL. In the second report, increased beta-cell expression of HK1 was detected by immunohistochemical staining of islets from several infants who underwent surgery for diazoxide-unresponsive congenital HI; these infants were also suggested to have HI because of a failure of HK1 silencing in beta cells. As described later, failure to silence expression of a “disallowed” beta-cell gene has also been suggested as the mechanism of exercise-induced HI: expression of the pyruvate plasma membrane transporter, MCT1.
Short Chain 3-Hydroxyacyl-Coa Dehydrogenase Hyperinsulinism
Short chain 3-hydroxyacyl-CoA dehydrogenase ([SCHAD], encoded by HADH on chromosome 4 at q22-26) is a mitochondrial fatty acid oxidation enzyme that catalyzes oxidation of short chain 3-hydroxyacyl-CoAs. Levels of SCHAD protein are relatively high in pancreatic beta cells, where it appears to negatively regulate the activity of GDH, the target of the gain-of-function mutations responsible for GDH-HI (the hyperammonemia HI syndrome). Homozygous inactivating mutations of the HADH gene are associated with increased beta-cell GDH activity and cause a form of HI similar to GDH-HI, but without hyperammonemia or specific brain defects. The systemic defect in fatty acid oxidation in SCHAD deficiency leads to accumulation of upstream substrates resulting in diagnostic elevations of urinary organic acids (3-hydroxyglutarate) and plasma acylcarnitines (3-hydroxybutyryl-carnitine). Patients with HI caused by SCHAD deficiency have responded well to treatment with diazoxide.
Hepatocyte Nuclear Factor 4a Hyperinsulinism
Hepatocyte nuclear factor 4a (HNF4A) is a transcription factor important for pancreatic beta-cell development and insulin secretion. Heterozygous inactivating mutations of HNF4A are a well-recognized cause of monogenic diabetes (MODY1). Recently, it was recognized that these same mutations also cause excessive insulin secretion in early life, as manifested by fetal macrosomia and persistent diazoxide-responsive neonatal hyperinsulinemic hypoglycemia. The HI is usually transient, lasting only a few weeks to a few months after birth; however, it may persist for several years into childhood. In the second and third decades, these children may develop the MODY1 form of diabetes, which is responsive to treatment with sulfonylureas. In most cases of HNF4A HI, birthweight has been above average, hypoglycemia occurred within the first days of life, and one parent had a history of MODY-type diabetes. Children with a specific HNF4A mutation, p.Arg76Trp, have additional manifestations, including renal Fanconi syndrome and hepatomegaly and elevated transaminases, suggesting that expression of the GLUT2 transporter in kidney tubules and in liver was impaired by the defect in HNF4A .
Hepatocyte Nuclear Factor 1A Hyperinsulinism
Hepatocyte nuclear factor 1a (HNF1A) is another transcription factor important for beta-cell development. Heterozygous inactivating mutations of HNF1A cause the most common form of monogenic diabetes, MODY3. Several cases of HNF1A mutations causing congenital HI have been reported, several of which also were associated with MODY diabetes in older patients. An interesting, unexplained discrepancy between HNF4A and HNF1A HI is that large for dates birthweight has only been associated with HNF4A , but not HNF1A mutations.
Uncoupling Protein 2 Hyperinsulinism
Uncoupling protein 2 (UCP2) transfers Krebs cycle intermediates out of the mitochondria and favors oxidation of amino acids versus glucose in beta cells, liver, and other tissues. Loss-of-function mutations of UCP2 may enhance ATP generation from glucose and consequently amplify insulin responses to glucose stimulation, sufficiently as to cause hyperinsulinemic hypoglycemia. Several cases have been reported, which have generally responded to treatment with diazoxide.
Exercise-Induced Hyperinsulinism
A total of 13 patients belonging to three families have been found with dominantly inherited exercise-induced hyperinsulinemic (EIHI) hypoglycemia following periods of intense anaerobic exercise. Clinical presentations have varied, with some cases having severe hypoglycemic episodes from infancy; some presenting with hypoglycemic syncope after exercise, as adolescents; and some only mildly affected, as adults. Most patients maintained normal glucose levels during prolonged fasting. Provocative testing, with brief, intense bicycle exercise, induced the expected rise in plasma lactate and pyruvate levels, but markedly increased plasma insulin concentrations for about 10 minutes after the exercise, causing hypoglycemia over the next 45 minutes. Intravenous infusion of pyruvate stimulated a similar rise in insulin, suggesting that the defect reflected an abnormal beta-cell responsiveness to pyruvate. Recurrent hypoglycemia was only partially preventable with diazoxide treatment.
Excessive insulin secretion in EIHI occurs because of mutations upstream of the promoter region SLC16A1 that allows the MCT1 monocarboxylate transporter to be expressed on the beta-cell plasma membrane. Normally, expression of MCT1 is “disallowed” in beta cells to prevent pyruvate and lactate from acting as insulin secretagogues.
Syndromic Forms of Congenital Hyperinsulinism
Beckwith-Wiedemann Syndrome
Beckwith-Wiedemann syndrome (BWS) is an overgrowth syndrome caused by embryonic mosaic mutations of an imprinted region on 11p, which is close to the location of the beta-cell KATP channel genes, SUR1 and Kir6.2 . The BWS locus includes growth-inhibiting genes expressed on the maternal chromosome (H19, P57) and growth-promoting genes ( IGF2 ) expressed on the paternal chromosome; expression of these genes is controlled by two imprinting control sites. Although BWS has been associated with HI for many years, recently it has been found that most cases of BWS with persistent HI are associated with paternal isodisomy for the 11p region (11pUPD overgrowth), which is the cause of around 20% of BWS cases. The histopathology in these cases shows islet cell overgrowth (adenomatosis), which may involve nearly the entire pancreas. Some cases are responsive to diazoxide, but hypoglycemia in some may not be controllable with diazoxide and may require more intensive medical management with octreotide and continuous feedings or pancreatectomy. Some cases of 11pUPD BWS also have a paternally transmitted recessive KATP channel mutation; these cases can have severe HI and unusually high glucose requirements. The possibility of 11pUPD BWS should be suspected in infants with HI who have features of BWS, such as hemihypertrophy, macroglossia, or umbilical hernia.
Kabuki Syndrome
Kabuki syndrome is associated with recognizable dysmorphic features including facial features resembling a Kabuki actor mask and caused by somatic mosaic mutations in either MLL2 ( KMT2D ) or KDM6A , two genes which regulate chromatin methylation. A proportion of infants with Kabuki syndrome have persistent hypoglycemia caused by HI, which may either be responsive or nonresponsive to treatment with diazoxide.
Turner Syndrome
Turner syndrome has been reported to be associated with congenital HI for several decades. A recent study found that the frequency of Turner syndrome in a large series of children with HI was increased ~ 50-fold, indicating that the risk of HI in Turner syndrome was around 1 in 1000 compared with the risk of HI in the general population of 1 in 40,000. Some of the Turner syndrome cases were diazoxide responsive, but others failed to respond to diazoxide and required pancreatectomy. The only consistent chromosomal anomaly in Turner syndrome girls with HI was a 45 XO karyotype, suggesting that loss of a gene on the X-chromosome was the cause of the HI. A possible candidate gene is KDM6A , a pseudoautosomal X-chromosome gene, because inactivating mutations of KDM6A are one of the causes of Kabuki syndrome in which there is also an increased frequency of HI.
Carbohydrate-Deficient Glycoprotein Hyperinsulinism
Congenital disorders of glycosylation (CDG) are caused by recessively inherited loss-of-function mutations in genes that regulate glycosylation of proteins. Most produce syndromic diseases affecting multiple organ systems, including brain, skeleton, liver, and kidney. Hypoglycemia resulting from HI has been reported in children with three of the identified defects of N-glycosylation: CDG Ia, Ib, and Id (OMIM IDs: 601785, 602579, and 601110). Most cases of CDG-HI have been identified in the newborn period, but some have been diagnosed later in infancy or early childhood. CDG-HI is usually suspected in a child with a dysmorphic facies and manifestations affecting multiple other organ systems, including—especially the brain, liver, gut, and skeleton. In all three types, cases have been reported in which hyperinsulinemic hypoglycemia was the presenting or dominant problem. The mechanism of excessive insulin production has not been determined.
Phosphomannomutase 2 (PMM2)-CDG (CDG-Ia) is the most common type of CDG and involves deficient activity of phosphomannomutase 2 resulting from mutations of PMM2 . Most patients have severe developmental delay, cerebellar hypoplasia, hypotonia, and seizures. Protein-losing enteropathy and liver disease contribute to failure to thrive. Deficient levels of antithrombin III can cause thromboses. Dysmorphic features are common and may be subtle or obvious, including unusual fat distribution and inverted nipples. Hyperinsulinemic hypoglycemia occurs in only a minority of patients, but can be mild or severe enough to warrant pancreatectomy.
Phosphomannose isomerase (MPI)-CDG (CDG-Ib) involves deficient activity of phosphomannose isomerase resulting from mutations of MPI . Unlike CDG-1a, the central nervous system is spared and hyperinsulinemic hypoglycemia is a common feature, presenting in the first days of life or later in the first year. Liver disease and protein-losing enteropathy are usually the dominant clinical problems. Some children have had cyclic vomiting. Clinical severity is variable, and mildly affected adults have been diagnosed. Oral mannose in doses of up to 150 mg/kg/day can correct most of the clinical abnormalities, making MPI-CDG the only CGD with a specific treatment.
ALD3-CDG (CDG-Id) involves deficient activity of mannosyltransferase 6 resulting from mutations of ALD3 . Clinical features resemble those of PMM2-CDG, with severe central nervous system damage. One case with severe neonatal hyperinsulinemic hypoglycemia has been reported, but whether it was a direct result of the CDG or a consequence of perinatal stress is unknown.
PMG-1 reversibly converts glucose-6-P to glucose-1-P and is required both for glycogen formation and degradation. Recessive inactivating mutations of PGM1 cause a syndrome of dysmorphic features, short stature, and fasting hyperketonemic hypoglycemia. In addition to abnormalities in protein glycosylation (hence the designation of CDG-It), hyperinsulinemic hypoketonemic hypoglycemia occurs when patients are fed. Deficiency of PGM1 impairs both the synthesis and breakdown of liver glycogen (hence the fasting hyperketonemic hypoglycemia and the labeling of the disorder as GSD type 14 ). However, deficiency of PGM1 in the beta cell appears to cause an exaggerated insulin response to a glucose load, leading to postprandial hyperinsulinemic hypoglycemia. The diagnosis can be made by screening tests for abnormalities in transferrin glycosylation. Treatment with diazoxide has not been effective in controlling postprandial hyperinsulinemic hypoglycemia, but low glycemic index feedings, uncooked cornstarch (UCS), or continuous low-dose glucose infusions have been more successful. Because the defect potentially impairs synthesis of galactose, treatment with galactose supplements have been tried with some success in correcting abnormalities of transferrin glycosylation.
AKT2 Hypoglycemia
Three children with hypoglycemia caused by gain-of-function mutations of AKT2 have been described. AKT2 is a serine/threonine kinase that is one of the downstream steps in postinsulin receptor signaling; loss of AKT2 activity is associated with insulin resistance, whereas activation of AKT2 causes an increase of insulin sensitivity. Beginning around 6 months of age, all of the reported cases had episodes of severe, symptomatic, hypoketotic hypoglycemia resembling HI, but associated with low levels of insulin, proinsulin, and FFA. Hypoglycemia was primarily postprandial and could be controlled with low dose continuous dextrose feedings. Other clinical features included large birth weight and postnatal asymmetric overgrowth of body or face. Each of the three unrelated patients had the same de novo mutation of AKT2 . The mutations were postzygotic and mosaic, which probably explains the localized nature of the overgrowth.
Phospatidylinositol-3-Kinase Hypoglycemia
Phospatidylinositol-3-Kinase (PI3K) is an enzyme step in the pathway of postinsulin receptor signaling upstream from AKT2. Postzygotic gain-of-function mutations of two PI3-kinase–related enzymes, PIK3CA and PIK3R2, were recently reported to cause a syndromic form of hypoketotic hypoglycemia with low insulin concentrations, similar to gain-of-function mutations of AKT2 described earlier. Features included asymmetric overgrowth, megalencephaly, and developmental delay. Diazoxide was ineffective and treatment with continuous dextrose at a low glucose infusion rate was needed to control the hypoglycemia. The hypoglycemia appears to be caused primarily by increased postinsulin receptor activity in the liver.
Hyperinsulinemic Hypoglycemia Associated With Insulin Receptor Defects
Inactivating mutations of the insulin receptor (INSR), which cause insulin resistance and diabetes mellitus, can also be associated with postprandial hyperinsulinemic hypoglycemia of varying severity. Severe defects in the INSR present in early infancy and childhood with dysmorphic features because of marked deficiency of adipose tissue (lipodystrophy) and, in order of severity, include Donohue syndrome (leprechaunism) (OMIM ID: 246200) and Rabson-Mendenhall syndrome (OMIM ID: 262190); milder defects may present later in childhood or early adulthood as type A insulin resistance (OMIM ID: 610549). More severe forms of insulin resistance may be caused by biallelic INSR mutations, and monoallelic defects may be associated with milder forms of insulin resistance; mutations in the INSR beta subunit tend to be milder than mutations in the alpha subunit. Plasma insulin levels are increased 10 to 100 times normal. Hypoglycemia in these patients is usually postprandial and has been suggested to be caused by delayed insulin clearance (a similar delay in insulin clearance has also been suggested to be responsible for postprandial hypoglycemia in autoimmune hypoglycemia caused by antiinsulin antibodies). Fasting hypoglycemia has been associated with the more severe insulin resistance seen in neonates and infants with Donohue syndrome and has been suggested to be caused by the very high insulin levels, having an insulin-like action by cross-reaction with the insulin-like growth factor-1 (IGF-1) receptor, although an earlier report demonstrated a pattern of accelerated hyperketonemic fasting adaptation, suggesting a possible impairment in hepatic glycogen storage. Infants with Donohue syndrome present in the newborn period and most die of infection in the first years of life. Features include intrauterine growth retardation, markedly diminished subcutaneous fat, typical facial appearance reminiscent of a leprechaun (“leprechaunism”), enlargement of breasts and clitoris or phallus, hypertrichosis, acanthosis nigricans, and postprandial hyperglycemia. Hypoglycemia, a few hours after meals, may be noted within the first days after birth and persists throughout life. Frequent feedings have been the mainstay of management for the hypoglycemia; diazoxide has generally not been effective.
Children with Rabson-Mendenhall syndrome are somewhat less severely affected than those with Donohue syndrome, but share many of the features, including reduced adipose tissue, acanthosis nigricans, hypertrichosis, and enlargement of the genitals in both males and females. Many of these children also have postprandial hypoglycemia. Diabetes tends to be progressive and insulin resistance can be severe. Adolescent females may have evidence of hyperandrogenism similar to polycystic ovarian syndrome. Death in childhood from diabetic ketoacidosis is common. Treatment with leptin (metreleptin) has been reported to modestly improve glucose control in some patients with Rabson-Mendenhall syndrome.
Postprandial (reactive) hypoglycemia has also been reported in adolescents with type A insulin resistance in which diabetes mellitus develops more gradually, sometimes as the adolescent reaches adulthood. Features may include reduced adipose tissue, acanthosis nigricans, and elevated triglyceride levels. Insulin resistance may lead to eventual beta-cell failure and insulin insufficiency.
Reactive hypoglycemia has also been reported in insulin-resistant conditions because of postreceptor disorders, and should be considered in other insulin resistance syndromes, such as congenital lipodystrophy (Berardinelli-Seip syndrome), because of recessive defects in AGPAT2 , BSCL2 , or PTRF .
Factitious Hyperinsulinism
Instances of factitious HI have been reported in infants and children, typically the result of a parent or other caregiver administering insulin or an insulin secretagogue. This form of child abuse is referred to as Munchausen by proxy and may cause needless pancreatic surgery and can be lethal. In most reported cases, the parent (often the mother) was a nurse or other medical professional or had access to insulin or sulfonylureas being used at home for treatment of diabetes. Typically, classic symptoms of HI are present, but can occur irregularly. As in other forms of HI, hypoglycemia is accompanied by suppression of plasma ketones and FFA and an abnormal positive response to glucagon. Fasting tests in the absence of the caregivers will be normal; however, extreme caution is required to ensure no interference by the suspected agent. An extremely high plasma insulin concentration (e.g., > 100 μU/mL) is sometimes a clue (especially likely to be found after administration of short- or intermediate-acting insulin, such as regular or isophane (NPH); in contrast, plasma insulin levels are usually only mildly elevated in HI from endogenous causes. Insulin administration has become increasingly difficult to detect by standard insulin assays because these can vary in their ability to detect insulin analogues (lispro, aspart, glargine, glulisine, detemir, degludec).
The most conclusive evidence of exogenous insulin is a discrepancy between elevated plasma insulin, but very low plasma C-peptide concentration, indicating that endogenous insulin production has been suppressed. Insulinomas may be associated with elevated plasma insulin, but values are rarely greater than 100 μU/mL and levels of C-peptide and proinsulin should be correspondingly elevated.
Surreptitious (or, more commonly, accidental) administration of long-acting oral hypoglycemic agents that induce insulin secretion, such as glyburide or other sulfonylureas, cause elevation of both insulin and C-peptide and may cause persistent hypoglycemia for 24 hours or longer. Routine toxicology screens of blood and urine may not detect sulfonylureas, but specific testing can be arranged if a specific drug is suspected.
If surreptitious insulin or oral hypoglycemic drug administration is suspected, specimens of plasma/serum, from the time of hypoglycemia, should be obtained for testing of plasma insulin and C-peptide concentrations and preserved for other tests that may become necessary to document the involvement of an exogenous agent. Consultation with the laboratory is helpful in interpreting and selecting appropriate tests. Treatment of the hypoglycemia with intravenous glucose is usually sufficient, but continuous intravenous glucagon may be needed in severe cases and diazoxide or octreotide may be helpful in cases of prolonged insulin hypersecretion.
Autoimmune Hypoglycemia
Two types of autoimmune hypoglycemia have been reported: one mediated by antibodies to insulin and the other by antibodies to the insulin receptor. Hypoglycemia caused by antiinsulin antibodies has been reported most often from Japan (“Hirata disease”), but cases have been described in both sexes, all ages, and from many regions of the world. Nearly all autoimmune hypoglycemia in infants and children have been of this type. The presumed mechanism of hypoglycemia is slow dissociation of insulin from antibodies during the postprandial or postabsorptive periods. The hypoglycemia is most often postprandial, but may be fasting, and has sometimes been severe enough to cause seizures or coma. Metabolic features are those of HI, with low plasma ketone and FFA levels. Assays of plasma insulin can give very high values that remain constantly high over time because of the interference by endogenous insulin binding antibodies (this may vary, depending on the assay method); in contrast, plasma C-peptide values may be suppressed, suggesting the possibility of surreptitious insulin or sulfonylurea administration. The disorder may be triggered by infections with common viruses or by exposure to drugs containing sulfhydryl or thiol groups, such as methimazole or lipoic acid, and appears to be associated with other autoimmune endocrine disorders, such as hyperthyroidism. In Japan, an association with the HLA allele HLA-DRB1*04:06 has been reported; a high incidence of HLA-DRB1*04:03 has been described in Europeans with the disorder. Among various treatments reported to improve the hypoglycemia have been courses of glucocorticoids, plasmapheresis, and intravenous immunoglobulin infusions. The disorder often resolves over time as autoantibody titers wane.
The second form of autoimmune hypoglycemia is caused by antibodies that bind to and activate the insulin receptor (analogous to thyroid-stimulating immunoglobulins in Graves disease). Childhood onset is rare, but cases as young as 8 months old have been seen. In children, there may be severe fasting hypoglycemia with very elevated rates of glucose utilization that is unresponsive to diazoxide or octreotide. In adults, the disorder has been associated with type B (autoimmune) insulin resistance, or a malignancy or severe inflammatory disorder. Some patients have had both antiinsulin and antireceptor antibodies. C-peptide and insulin levels are typically undetectable and diazoxide and octreotide are ineffective in controlling hypoglycemia. Treatment with glucocorticoids, plasmapheresis, or intravenous immunoglobulin may be considered.
Insulinoma
Insulinomas are the most common cause of acquired persistent hypoglycemia in adults and older children and may rarely occur in children as young as 2 years of age. Most often, insulinomas are solitary, small tumors, 0.5 to 1 cm in size, which are slow-growing and rarely become malignant or metastasize; multiple tumors and recurrent insulinomas may occur, especially in patients with multiple endocrine neoplasia type 1 (MEN1) syndrome because of dominant inactivating mutations in MENIN on 11q. Children with insulinomas present with recurrent episodes of symptomatic hypoglycemia, which are often initially mild and may go unrecognized for several months before severe manifestations, such as a seizure, occurs. The diagnosis of HI is most easily made with a fasting test to demonstrate hypoketonemic hypoglycemia, together with an inappropriately large glycemic response to glucagon. In younger children, especially, it may not be possible to distinguish clinically between an insulinoma and a genetic form of diffuse HI.
The majority of pediatric insulinomas appear to be associated with paternal 11p uniparental isodisomy, although this appears not to be so common in adult insulinomas. The frequency of genetic mutations in MENIN appears to be higher in children (42% of childhood insulinomas at Children’s Hospital of Philadelphia) than in adults with insulinomas (~ 8%). This is important, because the presence of multiple tumors at first surgery and recurrence of tumors is more likely in patients with MEN1 than in cases of sporadic insulinomas.
Initially, diazoxide may provide good control of hypoglycemia in patients with an insulinoma, especially in early stages, while the tumor is small; but diazoxide may become ineffective as the tumor grows. Surgical resection is the treatment of choice; therefore careful preoperative imaging to locate the lesion (or lesions) before surgery is important. In a series of eight pediatric cases, imaging was successful localizing the insulinoma in 40% by MRI, 40% by endoscopic ultrasound, 30% by computed tomography, and only 20% by abdominal ultrasound. Octreotide scan and F-DOPA PET scans occasionally provide additional diagnostic utility. At surgery, palpation and inspection and intraoperative ultrasound were generally successful in identifying and resecting the insulinoma. Patients with MEN1, caused by dominant mutations of MENIN , are at risk for other endocrine tumors, including functioning or nonfunctioning adenomas of the parathyroids, pituitary, and pancreas and require regular monitoring, as well as testing to identify affected family members.
Postprandial Hypoglycemia After Gastrointestinal Surgery (“Late Dumping Syndrome”)
Gastrointestinal hormones and signals—the enteroinsular axis—play an important role in regulating insulin secretion in response to meals. Disruption of this system by gastric surgery can result in hypoglycemia from excessive postprandial insulin release. In children, the most common cause is gastric surgery for gastroesophageal reflux (e.g., Nissen fundoplication); in adults, the most common cause is gastric bypass bariatric surgery for severe obesity. This “alimentary hypoglycemia” is exclusively postprandial (i.e., reactive), rather than fasting, and is caused by an amplified release of glucagon-like peptide 1 from the small intestine. The disorder is sometimes called late dumping syndrome to contrast it with early dumping syndrome , which is caused by rapid gastric emptying that induces osmotic shifts and symptoms, such as hypotension, sweating, and diarrhea, immediately after a feeding.
In young children, postprandial hypoglycemia after Nissen fundoplication may be accompanied by bowel symptoms of early dumping syndrome, but more often presents as isolated neuroglycopenic and autonomic symptoms 1 to 3 hours after meals. Hypoglycemia may be confirmed in association with symptoms after a regular feeding or can be elicited by a formal mixed meal or oral glucose tolerance test. Although the glucose tolerance is somewhat unphysiologic, and normal individuals may develop hypoglycemia 3 to 4 hours after a glucose load, the glucose tolerance test is preferred by some as a more standardized test. Patients with late dumping syndrome hypoglycemia have an unusually large rise in plasma insulin to as high as 200 to 300 μU/mL in the first 30 to 60 minutes after ingesting glucose, which is followed by hypoglycemia at 3 to 4 hours. There is often also a marked hyperglycemic spike shortly after glucose ingestion because of rapid gastric emptying; however, this does not always occur and is not the cause of the hypoglycemia.
Treatment of late dumping syndrome hypoglycemia may include dietary measures that slow gastric emptying and the release of free glucose into the small bowel, such as frequent small feedings of complex carbohydrates, small snacks between meals, avoidance of simple sugars, and providing fat and protein along with carbohydrates. In more severe cases, acarbose, an alpha-glucosidase inhibitor that slows digestion of starch and other complex carbohydrates, may be effective using doses between 25 and 100 mg, with each feeding. In some cases, continuous intragastric or intrajejunal tube feedings have been required.
Hyperinsulinemic Hypoglycemia as a Prodrome to the Onset of Diabetes Mellitus
Instances of fasting, but more commonly postprandial hypoglycemia, have occasionally been described before the onset of type 1 diabetes or during the “honeymoon” remission phase when insulin requirements are low. In the Diabetes Prevention Trial-Type 1, closely observed first- and second- degree relatives of patients with type 1 diabetes who were at increased risk of diabetes (i.e., individuals with prediabetes) were found to have chemical hypoglycemia detected by quarterly glucose profiles at a rate of 7.5 episodes per 100 person-years. Episodes of hypoglycemia, with documented blood glucose lower than 50 mg/dL, occurred at a rate of 2.6 episodes per 100-person years. There were no reported episodes of severe hypoglycemia. The mechanism most likely involves excessive second-phase insulin release as a consequence of a defective first-phase insulin response; other potential mechanisms have also been proposed, including delayed action of insulin, because of antiinsulin antibodies or insulin release secondary to inflammatory destruction of beta cells. A relationship between reactive hypoglycemia and onset of type 2 diabetes (“dysinsulinism”) was postulated as early as 1930, and widely recognized among diabetologists in the middle of the 20th century. Early type 2 diabetes is also often characterized by loss of the first-phase insulin response to food, resulting in higher glucose excursions, followed by lower glucose nadirs. In most cases of prodromal hypoglycemia in both type 1 and type 2 diabetes, plasma glucose does not reach levels low enough to cause neuroglycopenic symptoms. If necessary, treatment to avoid excessive postprandial hyperglycemia, such as low glycemic index meals, may be effective. In type 1 diabetes during the remission phase, raising the dose of insulin to reduce postprandial hyperglycemia may be helpful.
Hypoglycemia in Cystic Fibrosis Related Diabetes
In cystic fibrosis (CF) first-phase insulin secretion is impaired and glucagon secretion decreases with worsening glucose tolerance. Both fasting and reactive hypoglycemia during a routine glucose tolerance test is relatively common before the onset of cystic fibrosis–related diabetes (CFRD); however, the frequency of reactive hypoglycemia may not be more common than in individuals without CF. In one study, the occurrence of hypoglycemia following an oral glucose tolerance test (OGTT) was associated with a lower 10-year risk of progression to CFRD, as compared with those without hypoglycemia.
Nonislet Tumor Hypoglycemia
Certain noninsulin-secreting tumors are occasionally associated with paraneoplastic hypoglycemia. Most cases have involved large, malignant tumors of mesenchymal, epithelial, or hematopoietic origin. Cases in children are rare, but have been reported with neuroblastoma and Wilms tumor. The hypoglycemic episodes typically occur with fasting and are associated with inappropriately low plasma ketones and FFA, and with increased glucose utilization, which mimic insulin excess. Plasma levels of insulin, proinsulin, and C-peptide are low, and drugs which suppress insulin secretion (diazoxide, somatostatin) are ineffective, suggesting activation of insulin receptors by other circulating factors. In most cases, levels of IGF-2 have been found to be elevated and are postulated to cause hypoglycemia by cross-reaction with both insulin and IGF-1 receptors. IGF-2 is normally produced in the liver and limited amounts are secreted into the circulation bound to IGFBP-3 and the acid-labile subunit (ALS). Tumor-produced IGF-2 may be more active because of incomplete processing and less efficient binding to IGFBP-3 and ALS or may be structurally abnormal—a large molecular form called big IGF-2 . In addition to stimulation of the insulin receptor, stimulation of the IGF-2 receptor may downregulate GH secretion, resulting in low levels of IGFBP-3.
The demonstration of elevated plasma IGF-2 with suppression of both IGF-1 and insulin levels confirms tumor hypoglycemia of this variety. If the tumor cannot be completely removed, a possibly effective intervention is treatment with GH to raise IGFBP-3 levels and reduce the free IGF pool, thereby ameliorating hypoglycemia.
It should be noted that not all paraneoplastic hypoglycemia is mediated by IGF-2. Hypoglycemia associated with lactic acidemia, attributed to increased anaerobic glucose metabolism, has been reported in lymphomas and leukemias.
Glycogen Storage Diseases
The GSDs or glycogenoses comprise several inherited diseases caused by deficiencies of enzymes that regulate the synthesis or degradation of glycogen, resulting in increased storage of glycogen in several tissues, especially liver and muscle. Glycogen is the storage form of carbohydrate in humans: hepatic glycogen serves as a crucial glucose reservoir in the intervals between meals; glycogen in muscle and other tissues cannot be released as free glucose, but is used for endogenous needs. Hepatic glycogenolysis provides a large fraction of the glucose that enters the circulation, beginning within a few hours after eating, and continuing until the store of glycogen is depleted.
Glycogen is a highly branched polymer of glucose residues, most of which form straight chains linked by α-1,4-glycosidic bonds. Branches are created by α-1,6-glycosidic bonds, which occur on the average of once every 10 residues. After carbohydrates have been ingested, plasma glucose and insulin levels rise and new glycogen is synthesized.
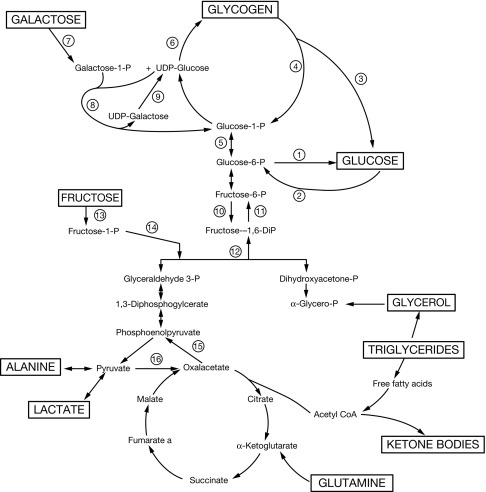
Glycogen synthesis and degradation in the liver follow distinct pathways that begin and end with glucose 1-phosphate (see Fig. 23.5 ). The liver is freely permeable to glucose, which is first converted to glucose 6-phosphate before it can enter one of several metabolic pathways. Glucose 6-phosphate can be reversibly converted to glucose-1-phosphate, which is the starting point for glycogen synthesis. Alternatively, glucose-6-phosphate can be hydrolyzed to glucose by glucose-6-phosphatase (G6Pase), or it can be metabolized via the glycolytic pathway to pyruvate and lactate, or via the pentose phosphate pathway, to ribose-5-phosphate, a precursor of nucleotide synthesis. Glycogen synthase catalyzes the formation of α-1,4-linkages. A branching enzyme forms the α-1,6-linkages that make glycogen a branched polymer.
Glycogen breakdown (glycogenolysis) requires the sequential interaction of several enzymes. First, hepatic phosphorylase successively cleaves the 1,4 links to within four glucosyl units of the branch point. Then 4-α-glucanotransferase exposes the 1,6-linked branch points by transferring three glucosyl residues to elsewhere on the glycogen molecule. Amylo-1,6-glucosidase, the debranching enzyme, then splits the 1,6-linked glucosyl units. Thus the sequential actions of phosphorylase and debrancher enzyme liberate the stored glucose units; the action of phosphorylase yields glucose 1-phosphate and the debranching enzyme liberates free glucose. During fasting, the debrancher enzyme mobilizes approximately 8% of hepatic glycogen as free glucose; the remainder requires activity of hepatic G6Pase.
The hallmark of the hepatic glycogenoses is fasting hypoglycemia. The types of hepatic glycogenoses, their specific enzyme deficiencies, affected tissues, their modes of inheritance, and the chromosomal localization of the relevant genes are shown in Table 23.3 . Table 23.4 summarizes the major biochemical characteristics of the hepatic glycogenoses (types 0, I, III, VI, and IX) that typically cause hypoglycemia.
| Disorder | Affected Tissue | Enzyme | Inheritance | Gene | Chromosome | Fasting Hypoglycemia |
|---|---|---|---|---|---|---|
| Type 0 GSD | Liver | Glycogen synthase | AR | GYS2 | 12p12.2 | Mild to moderate |
| Type Ia GSD | Liver, kidney, intestine | Glucose 6-phosphatase | AR | G6PC | 17q21 | Severe |
| Type Ib GSD | Liver | Glucose 6-phosphate transporter (T1) | AR | SLC37A | 11q23 | Severe |
| Type IIIa GSD | Liver, muscle, heart | Glycogen debranching enzyme | AR | AGL | 1p21 | Mild to moderate |
| Type IIIb GSD | Liver | Glycogen debranching enzyme | AR | AGL | 1p21 | Mild to moderate |
| Type IV GSD | Liver | Branching enzyme | AR | GBE | 3p12.2 | With liver failure |
| Type VI GSD | Liver | Glycogen phosphorylase | AR | PYGL | 14q21–22 | Mild |
| Type IXa GSD | Liver, erythrocytes, leukocytes | Liver isoform of α subunit of phosphorylase kinase | X-linked | PHKA2 | Xp22.1–p22.2 | Mild to moderate |
| Type IXb GSD | Liver muscle, erythrocytes, leukocytes | β subunit of liver and muscle phosphorylase kinase | AR | PHKB | 16q12–q13 | Mild to moderate |
| Type IXc GSD | Liver | Testis/liver isoform of γ subunit of phosphorylase kinase | AR | PHKG2 | 16p11–p12 | Mild |
| Type XI GSD | Liver, pancreas, intestine, and kidney | Glucose transporter 2 (GLUT2) | AR | SCL2A2 | 3q26.1-q26.3 | Mild |
| At Time of Hypoglycemia | Response to Oral Glucose | Response to Glucagon 4–8 Hours After a Meal a | Response to Glucagon 2 Hours After a Meal b | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Type | Triglyceride | Uric acid | Lactate | Glucose | Lactate | Glucose | Lactate | Glucose | Lactate |
| GSD-0 | N | N | N | ↑↑ | ↑↑ | 0-↑ | 0 | ↑ | ↓ |
| GSD-I | ↑↑↑ | ↑↑ | ↑↑↑ | ↑ | ↓↓ | 0 | ↑↑↑ | 0 | ↑↑ |
| GSD-III | ↑ | N | N | ↑ | ↑ | 0 | 0 | ↑ | 0 |
| GSD-VI, IX | 0-↑ | N | N | ↑ | ↑ | 0-↑ | 0 | ↑ | 0 |
a After a glucose-containing meal.
b See text to explain why there is a mild glycemic response early after a meal in the GSD types listed.
Glycogen Synthase Deficiency (Type 0 GSD)
Mutations in the glycogen synthase gene ( GYS2 ) on chromosome 12p12.2 cause a rare autosomal recessive disorder characterized by inability to store hepatic glycogen, resulting in markedly reduced hepatic glycogen content 4 to 6 hours after a meal (~ 0.5 g/100 g wet weight of liver as compared with 5 g/100 g in a normal child), but normal muscle glycogen content. Ingested glucose is preferentially converted to lactate. Symptoms of morning hypoglycemia appear when nocturnal feeding ceases. Type 0 GSD has a characteristic biochemical phenotype: fasting hypoglycemia and hyperketonemia alternating with transient hyperglycemia with glucosuria and hyperlacticacidemia during the daytime, especially after high-carbohydrate meals. During fasting hypoglycemia, plasma levels of the counterregulatory hormones are appropriately elevated, insulin levels are appropriately low, and ketones are elevated.
Unlike the other hepatic GSDs, the liver is not enlarged; therefore referral to glycogen synthase deficiency as a storage disease is a misnomer. Despite its rarity, the disorder should be considered in the differential diagnosis of “ketotic hypoglycemia.” After an overnight fast, oral glucose (1.75 g/kg) causes hyperglycemia and hyperlacticacidemia, whereas glucagon (0.03 mg/kg intramuscularly [IM]) typically has no appreciable effect on the plasma glucose level. Genetic testing for mutations in GYS2 is commercially available.
The goal of treatment is to prevent hypoglycemia and ketonemia during the night, and hyperglycemia and hyperlacticacidemia during the day. Fasting hypoglycemia and ketosis may be prevented by bedtime feedings of UCS, 1 to 1.5 g/kg. During illness, administration of a similar dose of cornstarch every 6 hours can be used to prevent hypoglycemia. During the day, patients are fed frequently (e.g., every 4 hours); because gluconeogenesis is intact, the diet should contain an increased amount of protein to provide substrate for gluconeogenesis and a decreased amount of carbohydrate (predominantly complex, low glycemic index carbohydrates) to minimize postprandial hyperglycemia and hyperlacticacidemia. This dietary regimen relieves symptoms, reverses the biochemical abnormalities, and improves growth.
Glucose-6-Phosphatase Deficiency (Type I GSD, Von Gierke Disease, Hepatorenal Glycogenosis)
First described in 1929, type I GSD is an autosomal recessive disorder caused by lack of activity of the hepatic enzyme G6Pase, either because of a deficiency of G6Pase itself or a deficiency of the transporter enzyme G6P translocase (G6PT). G6Pase catalyzes the final step in the production of glucose from glucose-6-phosphate. Deficiency of this enzyme impairs glucose production both from glycogenolysis and gluconeogenesis (see Fig. 23.5 ). Decreased production of glucose causes hypoglycemia in the intervals between meals, and increased production of lactate, uric acid, and triglycerides. Glycogen and triglycerides accumulate in the liver, resulting in marked hepatomegaly; glycogen also accumulates in the kidney and intestinal mucosa.
The G6Pase enzyme system is located in the endoplasmic reticulum (ER) membrane and consists of several subunits. The catalytic subunit, which converts glucose-6-phosphate to glucose, faces into the ER. Three transport systems transport the substrate, Glucose-6-phosphate, and the products, phosphate, inorganic orthophosphate, and glucose, across the ER membrane. glucose-6-phosphate transporter transports glucose-6-phosphate into and phosphate out of the ER; GLUT2 transports glucose out of the ER.
Approximately 80% of patients with GSD-I have deficient catalytic activity of the G6Pase system leading to type Ia GSD (GSD-Ia). Approximately 100 different mutations have been found in the G6Pase gene, G6PC , located on chromosome 17q21. Patients with GSD type Ib, which is caused by failure to transport glucose-6-phosphate into the lumen of the ER, have mutations (about 80 mutations have been described) in the G6PT gene, SLC37A4, on chromosome 11q23.
The estimated incidence of GSD-I is one in 100,000 births in the general population; its prevalence is one in 20,000 in Ashkenazi Jews. The presenting symptoms vary according to age. Symptomatic hypoglycemia may appear soon after birth; however, most patients are asymptomatic, as long as they receive frequent feedings that contain sufficient glucose to prevent hypoglycemia. Symptoms of hypoglycemia typically appear only when the interval between feedings increases, such as when the infant starts to sleep through the night or when an intercurrent illness disrupts normal feeding patterns. The condition may not be recognized until the child is several months old and an enlarged liver and protuberant abdomen are noted during a routine physical examination. Patients may present with hyperpnea (from lactic acidosis) and a low-grade fever without a demonstrable infection. Untreated patients may have a cushingoid appearance (a round cherubic or “doll-like” facies), growth failure, and delayed motor development. Social and cognitive development typically are not affected unless the infant suffers brain injury from recurrent severe hypoglycemia.
During infancy, the blood glucose concentration typically drops to less than 50 mg/dL within 3 to 4 hours of a feeding. Longer intervals of fasting cause even more severe hypoglycemia accompanied by hyperlacticacidemia and metabolic acidosis. Despite plasma glucose values in the range of 20 to 50 mg/dL, patients experience few autonomic or neuroglycopenic symptoms of hypoglycemia reflecting the brain’s ability to use lactate as an alternative fuel and the downregulation of autonomic responses after recurrent prolonged hypoglycemia. Serum insulin is appropriately suppressed and the counterregulatory hormones are elevated in a futile attempt to stimulate glycogenolysis and gluconeogenesis, resulting instead in lactic acidosis and exaggerated lipolysis, which causes marked hyperfattyacidemia. Fructose-1-6-bisphosphatase deficiency in infancy may resemble GSD I, with recurrent hypoglycemia and lactic acidosis (see Fig. 23.6 ); however, in contrast to GSD-I, glucagon administration elicits a brisk glycemic response in the recently fed state and genetic testing for FBP1 confirms the diagnosis (see Table 23.4 ).
The serum of untreated patients may be lipemic, with extremely high triglyceride concentrations (typically > 500 mg/dL) and moderately increased levels of phospholipids and total and low-density lipoprotein-cholesterol; the high-density lipoprotein-cholesterol concentration is low. High triglyceride levels are caused by increased hepatic synthesis (mobilization of FFA from adipose tissue in response to hypoglycemia), impaired ketogenesis, and reduced clearance of triglycerides, secondary to decreased lipoprotein lipase activity. Eruptive xanthomata may appear on the extensor surfaces of the extremities and on the buttocks and severe hypertriglyceridemia (> 1000 mg/dL) may cause acute pancreatitis.
Untreated or poorly controlled GSD I is associated with marked elevations of plasma uric acid. The hyperuricemia is caused by increased degradation of adenosine monophosphate (AMP) to inosine and uric acid because of deinhibition of AMP deaminase as a consequence of accumulation of glucose-6-phosphate and lowering of both ATP and inorganic phosphate in hepatocytes. A similar mechanism causes hyperuricemia in fructose-1,6-bisphosphatase deficiency (see later section on Genetic Disorders of Gluconeogenesis) because of accumulation of fructose-1,6-phosphate and is the reason fructose is not used in intravenous fluids.
Although symptomatic hypoglycemia becomes less severe with increasing age, nonetheless, without adequate therapy growth is stunted, and puberty is delayed. However, when continuous glucose therapy is started early in life and long-term good metabolic control is maintained, patients can grow and develop normally.
Impaired platelet function causes a bleeding tendency manifested as recurrent epistaxes or oozing after dental or other surgery. Reduced platelet adhesiveness, abnormal platelet aggregation, and impaired release of adenosine diphosphate (ADP) in response to collagen and epinephrine have been observed. The defects in platelet function are secondary to the systemic metabolic abnormalities and are corrected by improving the metabolic state.
Anemia is common in children with GSD I. The etiology of the anemia is multifactorial and includes iron and other nutritional deficiencies, chronic lactic acidosis, blood loss from menorrhagia, end-stage renal disease (ESRD), and enterocolitis (in patients with GSD Ib). Patients with liver adenomas may have iron refractory anemia secondary to aberrant hepcidin expression. Intestinal symptoms are usually not a prominent feature but can include diarrhea.
Kidney enlargement is readily demonstrated by ultrasonography. Deficiency of G6Pase leads to glycogen deposition in the kidneys and disturbs the metabolism of renal tubular cells, resulting in a relative energy deficiency. Increased renal blood flow and glomerular filtration rate (GFR) may be a compensatory mechanism for the intracellular energy deficit. Renal manifestations include proximal and distal renal tubular dysfunction, as well as glomerular injury that can lead to ESRD. Proximal tubular dysfunction (glucosuria, phosphaturia, hypokalemia, and a generalized aminoaciduria) is reversible when biochemical control of the disease improves. Some patients have a distal renal tubular acidification defect associated with hypocitraturia and hypercalciuria, which predisposes to renal calculi and nephrocalcinosis. Increased urinary albumin excretion may be observed in adolescents. Progressive severe renal injury with proteinuria, hypertension, and decreased creatinine clearance because of focal segmental glomerulosclerosis and interstitial fibrosis, may be seen in young adults. Patients with persistently elevated blood lactate, serum lipid, and uric acid concentrations appear to be at increased risk of developing nephropathy. Normalization of metabolic parameters decreases proteinuria, and optimal therapy instituted at or before age 1 year may delay, prevent, or slow the progression of renal disease.
Development of hepatic adenomas is a common complication, occurring in up to 75% of patients by the time they reach adulthood. Although adenomas are usually first observed in the second and third decades of life, they may appear before puberty. Adenomas may hemorrhage or become malignant hepatocellular carcinomas. Ultrasonography is the preferred method of screening for hepatic adenomas. When malignancy is suspected, MRI and measurement of serum alpha-fetoprotein should be performed.
Radiographic studies have demonstrated osteopenia, and pathologic studies have shown osteoporosis, without evidence of abnormalities in calcium, phosphate, parathyroid, or vitamin D metabolism. Bone mineral content is decreased compared with age-matched normal children. Endocrine and metabolic disturbances, including hypercortisolemia, resistance to GH, delayed puberty, and lactic acidosis, may account for decreased bone mineralization.
Menstrual irregularities and hirsutism caused by hyperandrogenism are uncommon; however, in all types of hepatic GSDs, ultrasonography has shown a high prevalence of morphologically polycystic ovaries (even in prepubertal children), the clinical significance of which is still unclear.
Although the heart itself is not affected in GSD I (unlike GSD III), hypertension may develop in association with renal disease. Pulmonary hypertension presenting in the second or third decade of life and leading to death from progressive heart failure has also been described.
Patients with GSD Ib have symptoms similar to GSD Ia, but with the addition of either constant or cyclic neutropenia of variable severity, ranging from mild to complete agranulocytosis, and is associated with recurrent bacterial infections. Neutropenia is a consequence of disturbed myeloid maturation and is accompanied by functional defects of circulating neutrophils and monocytes. A recent case series suggests that vitamin E supplementation may improve neutropenia and reduce infection rates. Patients frequently develop an inflammatory bowel disease resembling Crohn disease, which is responsive to treatment with granulocyte colony-stimulating factor (G-CSF). Children with GSD Ib are prone to oral complications (recurrent mucosal ulceration, gingivitis, and rapidly progressive periodontal disease), painful perianal abscesses, and have an increased prevalence of thyroid autoimmunity and primary hypothyroidism.
Diagnostic Studies
During infancy, plasma glucose typically decreases to less than 50 mg/dL within 3 to 4 hours of a feeding and is accompanied by hyperlacticacidemia and metabolic acidosis. The serum may be cloudy or milky, with very high triglyceride and moderately increased levels of cholesterol. Serum uric acid, aspartate aminotransferase, and alanine aminotransferase levels are usually increased. The simplest method of determining the probable nature of the enzymatic deficiency in a child suspected of having a GSD I is to obtain serial blood samples for measurement of metabolites (glucose, lactate, FFA, ketones, and uric acid) for up to 6 hours (or until the plasma glucose decreases to ≤ 50 mg/dL), following oral glucose (1.75 g/kg), or 30 minutes after stopping a continuous overnight intravenous or intragastric glucose infusion. Hypoglycemia, together with elevated blood lactate levels, combined with an abnormal fed glucagon stimulation test, clinches the diagnosis. In the normal child, a 2-hour postprandial (fed) glucagon stimulation test causes the plasma glucose to increase by 30 mg/dL or more within 30 minutes without a change in lactate concentration, whereas in GSD I, the glucose response is minimal and there is a marked increase in lactate. Because administration of glucagon can lead to pronounced worsening of metabolic acidosis and decompensation in GSD I, it is no longer recommended in the diagnostic evaluation of suspected GSD I. Mutation analysis of G6PC (GSD Ia) and SLC37A4 (GSD Ib) genes is recommended to confirm type I GSD. Because GSD Ia is more common, unless neutropenia is present, complete G6PC sequencing is recommended first. Note that although neutropenia suggests GSD Ib, it has also been occasionally seen in GSD Ia and the neutrophil count can be normal in patients with GSD Ib in the first years of life. A liver biopsy for measurement of G6Pase activity may be necessary in the rare patient who does not have an identifiable gene mutation. In most individuals with GSD Ia, G6Pase enzyme activity is < 10% of normal. G6Pase activity must be assayed in both intact and fully disrupted microsomes, as enzyme activity normalizes when freezing disrupts the integrity of the ER. For this reason, G6Pase enzyme activity on snap-frozen liver biopsy tissue will not detect GSD Ib, and fresh (unfrozen) liver biopsy tissue is needed to assay G6PT activity accurately. The glycogen itself has a normal appearance. Ultrasound of the abdomen shows hepatomegaly and nephromegaly without splenomegaly. Nephromegaly is characteristic of GSD I and is not a feature of the other GSDs.
Treatment consists of providing a continuous dietary source of glucose to prevent the plasma glucose from falling below the threshold for glucose counterregulation. When hypoglycemia is prevented by providing an appropriate amount of glucose throughout the day and night, the biochemical abnormalities are ameliorated, liver size decreases, the bleeding tendency is reversed, and growth improves. Recommended biochemical targets: plasma glucose greater than 70 to 75 mg/dL, triglycerides less than 500 mg/dL, and uric acid less than 7.5 mg/dL.
Various methods may be used to provide a continuous source of glucose at a rate sufficient to satisfy glucose requirements in the intervals between meals: intravenously, via the gastrointestinal tract by intragastric infusion (either nasogastric or gastrostomy tube), or by use of low glycemic index foods. Of the latter, UCS has the most suitable properties described to date. The minimum amount of glucose required in the basal state may be obtained by using the formula for calculating the basal glucose production rate: y = 0.0014 3 − 0.214x 2 + 10.411x − 9.084, where y = mg glucose per minute and x = ideal body weight in kilograms. The amounts and/or schedule of glucose or UCS administered are modified, if necessary, based on the results of clinical and biochemical monitoring.
Infants with GSD I usually require feeds every 2 to 3 hours of a formula that does not contain either lactose or sucrose. The formula must contain a polymer of glucose (corn syrup solids, maltodextrins) that will yield an amount of glucose equal to the calculated glucose production rate. If nighttime feedings are challenging, continuous overnight feedings, using the same formula, may be given via nasogastric or gastrostomy tube controlled by an infusion pump.
Orally administered UCS acts as an intestinal reservoir of glucose that is slowly absorbed into the circulation. In many centers, UCS has replaced frequent daytime feedings of glucose or glucose polymers and overnight continuous intragastric infusion of glucose. It has been used successfully in infants as young as 8 months of age. The UCS is given in a slurry of water or an artificially sweetened beverage (e.g., Kool-Aid) or in formula for infants, at 3- to 5-hour intervals during the day, and at 4- to 6-hour intervals overnight. The amount given is determined by multiplying the time interval between feedings by the calculated hourly glucose requirement for ideal body weight. One tablespoon (8 g) of UCS contains 7.3 g of carbohydrate. The optimum schedule and amounts of intermittent UCS feedings for patients of different ages is determined empirically by metabolic monitoring to ensure that the biochemical goals are being achieved.
Extended Release Waxy Maize Cornstarch (Glycosade, Vitaflo, International Ltd., Liverpool, England) was approved in England in 2009 for the management of GSD I and was released in the United States as a medical food in 2012. Efficacy studies in children as young as 5 years old have shown prolongation of fasting tolerance from an average of 4.1 to 7.8 hours, using extended release cornstarch as compared with standard UCS. Gastrointestinal intolerance and exacerbation of inflammatory disease are common side effects of the extended release formulation.
When adequate exogenous glucose is provided, significant hyperuricemia and hyperlipidemia are usually restored to near normal. If severe hyperuricemia persists, allopurinol, a xanthine oxidase inhibitor (5–10 mg/kg/day as a single daily dose or divided q12 hours), effectively lowers serum uric acid to normal levels. Lipid-lowering agents (gemfibrozil or fenofibrate) are indicated when persistent severe hyperlipidemia, despite optimal glucose therapy, poses a significant risk of acute pancreatitis.
Dietary fat should be restricted to less than 20% of the total energy intake, equally distributed among monounsaturated, polyunsaturated, and saturated fats. Dietary cholesterol is restricted to less than 300 mg/day. Carbohydrate typically provides 60% to 65% of the daily calories. Of the total daily calories, 30% to 45% is prescribed (both the amount and schedule) in the form of UCS. Most of the remaining dietary carbohydrate should, ideally, be low glycemic index starches. With the glucose requirements prescribed, the total caloric intake is determined by the child’s appetite, as long as the rate of weight gain is not excessive. The dietitian must ensure that the patient is consuming an adequate amount of protein, fat, minerals, and vitamins to support optimal growth. When adequate glucose is prescribed to maintain normoglycemia, milk products and fruit, despite their content of galactose and fructose, respectively, may be used sparingly to supply essential nutrients, minerals, and vitamins.
Renal disease
Angiotensin converting enzyme (ACE) inhibitors and angiotensin receptor blockers have been reported to improve GFR in patients with hyperfiltration. Thiazide diuretics and oral citrate can help prevent nephrocalcinosis and renal calculi. Loop diuretics should be avoided because of the risk of hypercalciuria.
Anemia
Iron, vitamin B 12 , and folic acid intake should be optimized. Patients with hepatic adenomas may develop iron refractory anemia caused by aberrant tumor expression of hepcidin, a key regulator of iron metabolism. Erythropoietin supplementation may be required in patients with ESRD.
Neutropenia
G-CSF is efficacious for treatment of neutropenia and infections in patients with GSD-Ib; monitoring for splenomegaly should be performed because it is the most serious complication of G-CSF therapy in this population.
Monitoring
Frequent plasma glucose monitoring before meals, before cornstarch administration, and before and after exercise is essential to establish good metabolic control. If the cornstarch dose is changed, plasma glucose should also be monitored frequently after cornstarch administration to establish the effective duration of action. Clinical studies of continuous glucose sensors, in both adults and children, show that they can be used safely and reliably in patients with GSD I, and can help reduce duration of hypoglycemia, as well as detect asymptomatic hypoglycemia.
Routine monitoring should include: complete blood count with differential white cell count, serum uric acid, triglycerides, and cholesterol. Triglyceride concentrations are considered the most useful parameter of metabolic control, given the rapid fluctuations in the levels of plasma glucose, lactate, and transaminases. Significantly increased adenoma progression has been reported in patients with mean triglyceride greater than 500 mg/dL.
The American College of Medical Genetics and Genomics has provided guidelines for screening patients with GSD I for complications. These include measurement of serum creatinine, urea, electrolytes, calcium, phosphate, liver function tests, protein, and albumin every 6 months. Urine microalbumin, protein, creatinine, calcium, and citrate should be checked every year from 0 to 5 years of age and every 6 months if older than 5 years of age. Creatinine clearance (GFR estimate) should be performed yearly for those older than 5 years of age. An abdominal ultrasound examination to evaluate liver, kidneys, spleen, and ovaries should be performed annually if younger than 10 years of age, and every 6 months after age 10 years. Bone densitometry should be obtained every 1 to 2 years for those older than 5 years. Periodic echocardiography (ECHO) and electrocardiogram (ECG) should be performed to screen for pulmonary hypertension starting at 10 years of age and repeated at least every 3 years.
A clinical study to evaluate the efficacy and safety of gene therapy to replace hepatic G6Pase in human subjects, using adeno-associated virus carrying the human G6PC gene, is currently in progress.
Amylo-1,6-Glucosidase Deficiency (Type III GSD, Debranching Enzyme Deficiency, Limit Dextrinosis, Cori Disease, Forbes Disease)
Type III GSD is caused by deficient activity of the glycogen debranching enzyme (GDE) (see Fig. 23.5 ), leading to impaired glycogen degradation, reduced glycogenolysis, and accumulation of abnormal glycogen (limit dextrin). It is an autosomal recessive disease resulting from mutations of the AGL gene on chromosome 1p21, which encodes four GDE isoforms. Isoform 1, the most widely distributed and the predominant form in the liver, is defective in GSD-IIIa. Isoforms 2, 3, and 4 are exclusively located in skeletal and cardiac muscle. GDE has two enzymatic functions: a transferase that exposes the 1,6-linkage and amylo-1,6-glucosidase, which produces free glucose. Without GDE, glycogen breakdown cannot proceed past the outermost branch points, resulting in limit dextrin, an abnormal form of glycogen with short side chains. Only the outer 5% to 10% of glucose residues can be released by phosphorylase. In the United States, 80% to 85% of patients with GSD-III lack GDE activity in both liver and muscle (GSD-IIIa), 15% lack GDE activity in the liver alone (GSD-IIIb); rarely, patients lack activity in muscle alone (GSD-IIIc). In contrast, in Israel the majority of patients lack enzyme activity only in the liver (GSD-IIIb).
Clinical and enzymatic variability is a feature of GDE deficiency. During infancy and childhood, the presentation may be indistinguishable from GSD-I. Hepatomegaly, fasting hypoglycemia with ketosis, hyperlipidemia, and growth retardation are the predominant features. About 70% of patients have muscle weakness, but this is usually not clinically significant in childhood.
Because small amounts of glucose can be produced from 1,4-segments beyond the outermost glycogen branch points and from gluconeogenesis, patients with GDE deficiency may be able to tolerate longer periods of fasting, and hypoglycemia usually is less severe than in those with G6Pase deficiency. Infants with GSD-III may be asymptomatic on their usual frequent feeding schedules and typically do not become as severely ill with infections and other stresses that disrupt feeding as do children with GSD-I.
Infants with GSD-III develop fasting hyperketonemia as a result of an accelerated transition to the fasting state. Blood lactate and uric acid levels are normal, because the gluconeogenic pathway is intact and hepatic glycolysis is not increased (see Fig. 23.5 ). Hyperlipidemia is less severe in GSD-III than in GSD-I. The presenting clinical finding may be hepatomegaly and growth failure. An enlarged spleen may be seen at 4 to 6 years of age in patients who develop hepatic fibrosis. The kidneys are not large and renal dysfunction does not occur. Untreated infants and children grow slowly and puberty is delayed. Patients who lack GDE in muscle usually have minimal, clinically insignificant muscle weakness during childhood. With the exception of myopathy, symptoms and signs characteristically ameliorate with increasing age. Myopathy, however, usually becomes prominent in the third or fourth decade of life, manifesting as slowly progressive muscle weakness involving the proximal muscles. In some patients, the small muscles of the hands are also affected. Cardiac involvement is common as a result of limit dextrin accumulation in the heart causing a cardiomyopathy that resembles idiopathic hypertrophic cardiomyopathy. Significant concentric left ventricular hypertrophy usually develops after puberty and manifests as ventricular hypertrophy on ECG and increased left ventricular mass and wall thickness on an echocardiogram. Ventricular changes are more commonly seen in patients with GSD-IIIa, but have also been reported in patients with GSD-IIIb. Symptomatic cardiomyopathy may occur and sudden death has been attributed to cardiac arrhythmias; glycogen accumulation in the cardiac conduction system has been observed at autopsy.
The size of the liver tends to decrease during puberty. Biopsy usually shows hepatic fibrosis, and some adult patients develop cirrhosis. Hepatic adenomas have been found in 4% to 25% of patients, compared with 22% to 78% of GSD-I patients. The development of hepatocellular carcinoma is rare, but has been described in patients more than 30 years old.
Typical biochemical features are fasting hypoglycemia with ketosis (i.e., ketotic hypoglycemia), but without elevation of blood lactate or serum uric acid concentrations (see Fig. 23.6 ). Hypertriglyceridemia and hypercholesterolemia occur in approximately two-thirds and one-third of patients, respectively. Hypertriglyceridemia is most commonly found in younger children (< 3 yearsold) and does not reach levels high enough to cause pancreatitis. Liver transaminases are consistently elevated in children (at least twice the upper limit of normal, and often > 500 IU/L), but decline at puberty and may be normal in adults. Creatine kinase (CK) is often markedly elevated in patients with GSD-IIIa.
After an overnight fast, plasma glucose and lactate concentrations do not increase after administration of glucagon (see Table 23.4 ). However, when the test is performed 2 hours after a high-carbohydrate meal (before the outer branches of glycogen are degraded), a glycemic response can be elicited. Diagnosis of GSD-III is made by mutation analysis of the AGL gene or, alternatively, by liver and/or muscle biopsy. Biopsy results show a 3- to 5-fold elevation of structurally abnormal (highly branched) glycogen content in the involved tissue. Definitive subtyping of GSD-III requires a biopsy of both liver and muscle. Although muscle involvement can be inferred from the presence of high levels of serum CK, a normal level does not rule out muscle enzyme deficiency. At the time of diagnosis, muscle biopsy is the only way to predict accurately whether skeletal muscle disease or heart muscle involvement is likely to develop in the future, unless a GSD-IIIb specific mutation is identified.
Because only a limited amount of glucose can be mobilized from glycogen, hypoglycemia develops during an overnight fast in infancy and early childhood. This occurs despite increased gluconeogenesis and enhanced hepatic uptake of gluconeogenic amino acids, which results in low plasma levels of several amino acids, such as alanine. As in GSD-I, continuous provision of an adequate amount of glucose, using UCS, combined with a normal intake of total calories, protein, and other nutrients, corrects the clinical and biochemical disorder and restores normal growth. UCS 1 to 1.75 g/kg at 6-hour intervals (e.g., at midnight, 6 am , etc.) maintains normoglycemia, increases growth velocity, and decreases serum aminotransferase concentrations. Infants less than 12 months old may not produce sufficient amylase to digest UCS and experience bloating, flatulence, and diarrhea. If these side effects occur, a slow rate of introduction of UCS or addition of pancrelipase (which contains amylase) may improve tolerance. UCS can be mixed in any beverage, although addition to milk or yogurt is preferable, as it adds protein and fat. Addition of whey protein is recommended to prolong normoglycemia. Excessive UCS should be avoided because it leads to excess glycogen deposition and weight gain. For patients who have significant growth retardation and myopathy, continuous nocturnal feeding of a nutrient mixture composed of glucose, glucose oligosaccharides, and amino acids combined with intermittent feedings during the day of meals with a high protein content may be beneficial. Adolescents and adults with GSD IIIa should aim for a high-protein (25% of total calories) and low complex carbohydrate (< 50% of total calories) diet and should avoid simple sugars and fasting. A high-protein diet may improve muscle function by enhancing muscle protein synthesis and reducing glycogen storage. It has also been reported that a high-protein diet (~ 30% of total calories) may reverse and possibly prevent cardiomyopathy. Patients with GSD-IIIb may be able to transition to a regular well-balanced diet.
Plasma glucose monitoring should be performed before meals or UCS, before bedtime and breakfast, with addition of extra monitoring during times of illness. Liver function tests should be performed every 6 months. As in patients with GSD-I, annual hepatic ultrasound examinations should be performed to screen for hepatic adenomas. MRI of the liver should be performed every 6 to 12 months in older patients and in children, when an ultrasound examination shows an adenoma has increased in size. Routine ECG and ECHO should be performed to evaluate for dysrhythmias, ventricular hypertrophy, and systolic or diastolic dysfunction. Cardiac ECHO should be done every 12 to 24 months in patients with GSD-IIIa and every 5 years in those with GSD-IIIb. ECG should be done every other year in patients with GSD-IIIa. No exercise restrictions are recommended for patients with GSD-III, unless they have significant cardiac disease. Exercise helps prevent worsening myopathy and low bone mineral density. Physical therapy evaluation is recommended every 6 months.
Hepatic Phosphorylase Complex Deficiency (Type VI GSD, Hepatic Phosphorylase Deficiency, Hers Disease; Type IX GSD, Phosphorylase Kinase Deficiency)
Hepatic phosphorylase, the rate-limiting enzyme of glycogenolysis, is activated by a cascade of enzymatic reactions triggered by glucagon and epinephrine. First, adenylate cyclase catalyzes the formation of cyclic adenosine monophosphate (cAMP), which then activates a cAMP-dependent protein kinase. Protein kinase then phosphorylates a phosphorylase kinase (PHK), which converts inactive hepatic phosphorylase to its active form. Active phosphorylase hydrolyzes the α-1,4-linkages and mobilizes glucose from glycogen (see Fig. 23.5 ).
The GSDs caused by a reduction in liver phosphorylase activity are a heterogeneous group of disorders (see Table 23.3 ). Hepatic PHK or type IX GSD, which occurs in approximately one in 100,000 births and accounts for about 25% of all cases of GSDs is the most common form; whereas, deficiency of hepatic phosphorylase (type VI GSD) itself is rare.
PHK of liver and muscle is a complex enzyme consisting of four subunits: α, β, γ, δ. The enzyme is regulated by phosphorylation of specific serine residues of the α and β subunits and by calcium through the δ subunit, a member of the calmodulin family. The γ subunit is catalytically active. Mutations in three different genes of PHK subunits ( PHKA2, PHKB, and PHKG2 ) can result in deficient activity of hepatic phosphorylase. GSD IXa, because of mutations in PHKA2 , is the most common variant (see Table 23.3 ).
GSD IXa is an X-linked disorder restricted to boys. Patients seldom have symptomatic hypoglycemia during infancy, unless they fast for a prolonged period. They can develop hyperketonemia similar to, but usually milder than, that seen in type III GSD. Metabolic acidosis is rare. The disorder is usually discovered when an enlarged liver and protuberant abdomen are noted during a physical examination. Physical growth is usually impaired, and motor development may be delayed as a consequence of hypotonia. With increasing age, clinical and biochemical abnormalities gradually ameliorate, and catch-up growth may occur; most adult patients are asymptomatic. GSD IXc, because of PHKG2 mutations, tends to have a more severe phenotype that includes liver fibrosis and cirrhosis in childhood. Recent reports indicate that liver fibrosis may also occur in GSD VI and IXa. Mild cardiomyopathy has been identified using ECHO on long-term follow-up of patients with GSD VI and IXb.
Hypoglycemia is unusual and blood lactate and uric acid levels are normal. Mild hypertriglyceridemia, hypercholesterolemia, and elevated transaminase levels may be present. Ketonemia occurs with fasting. Functional tests are not especially useful in evaluating these patients. After an overnight fast, blood lactate level is normal, and administration of glucagon elicits a brisk glycemic response, without a rise in the blood lactate concentration. The glycemic response to glucagon cannot be used to distinguish between phosphorylase kinase deficiency and lack of phosphorylase itself. Thus the definitive diagnosis of type IX GSD requires genetic testing or determination of phosphorylase kinase activity in erythrocytes or leukocytes. The diagnosis of type VI GSD can also be established by genetic testing or, alternatively, by assaying the activity of phosphorylase in purified blood cell fractions, and usually does not require a liver biopsy. Muscle phosphorylase activity is normal; muscle histology and glycogen content are normal.
In the past, specific treatment other than avoidance of prolonged fasting was considered unnecessary. Recent research, however, suggests that therapy with a high-protein diet and UCS in patients with GSD IX improves linear growth, general wellbeing, decreases hepatomegaly, and may be associated with regression of ultrasound findings of liver fibrosis. UCS, 2 g/kg at bedtime, typically prevents hypoglycemia and ketosis in these patients.
Plasma glucose and ketone monitoring should be done routinely. Growth and pubertal progression should be carefully monitored. Annual liver ultrasound examinations should be performed starting at age 5 years. Because there is increased risk of osteoporosis, bone densitometry is recommended once growth is complete.
Glycogen Branching Enzyme Deficiency (Type IV GSD, Andersen Disease)
There is minimal impairment of glycogenolysis and gluconeogenesis is normal; accordingly, hypoglycemia is rare; however, fasting hypoglycemia may occur when severe cirrhosis develops.
Fanconi-Bickel Syndrome (Type XI GSD)
GSD XI is a rare autosomal recessive disorder characterized by hepatorenal glycogen accumulation caused by deficiency of the facilitative GLUT2 that mediates the bidirectional transport of glucose and galactose in hepatocytes, pancreatic beta cells, enterocytes, and in the proximal tubules of the kidney.
Patients usually present in infancy with failure to thrive, chronic diarrhea from carbohydrate malabsorption, hepatomegaly, and fasting hypoglycemia, as the interval between feeds increases. A general renal proximal tubulopathy with severe glucosuria and hypophosphatemic rickets are characteristic features. Older patients have a moon-shaped face, a protuberant abdomen, short stature, and delayed puberty. Kidneys are enlarged (detectable by ultrasound) as a result of glycogen accumulation. Clinical findings are of variable severity.
Fasting ketotic hypoglycemia and postprandial hyperglycemia and hypergalactosemia (caused by impaired hepatic uptake of the two sugars) are characteristic biochemical features. Decreased insulin secretion (because of an impairment of beta-cell glucose sensing) may also contribute to impaired hepatic glucose uptake and postprandial hyperglycemia. Other laboratory findings include hypergalactosemia, glucosuria, renal bicarbonate wasting, proteinuria, phosphaturia, generalized aminoaciduria, and elevated serum alkaline phosphatase level. Mutation analysis of the GLUT2 gene confirms the diagnosis.
Frequent feeds using slowly absorbed carbohydrates and restriction of galactose are recommended. Supplementation with UCS has a beneficial effect on metabolic control and growth. Fructose metabolism is not affected; therefore this monosaccharide may be used as an alternative carbohydrate source. Water and electrolytes must be replaced. Alkali may be necessary to compensate for renal tubular acidosis, and hypophosphatemic rickets requires supplemental phosphate and vitamin D.
Ketotic Hypoglycemia
Ketotic hypoglycemia has been recognized for nearly a century as the most common type of childhood hypoglycemia between 1 to 6 years of age, with a well-characterized presentation and course, but an incompletely understood etiology. Usually, this condition presents as recurrent episodes of morning hypoglycemia in the second or third year of life—but onset as early as 6 months has been reported. The condition usually remits spontaneously by the age of 8 to 9 years. The classic history is of a child, often a male with a history of low birthweight, who has eaten poorly the previous day or misses an evening meal, is difficult to rouse from sleep the next morning, and displays neuroglycopenic symptoms that may range from lethargy to seizure. Hypoglycemic episodes are especially likely to occur during an illness, when food intake is limited. Treatment to avoid prolonged overnight fasting usually limits recurrent attacks to less than one to two a year.
At the time of hypoglycemia, high levels of ketones are found in plasma and urine, and plasma insulin concentrations are suppressed, indicating that HI is not responsible for the hypoglycemia. It remains unsettled whether these children display an accelerated, but qualitatively normal, metabolic response to fasting (i.e., the lower end of the normal distribution of fasting tolerance, “accelerated starvation”) or whether ketotic hypoglycemia represents a heterogeneous group of disorders of limited substrate availability awaiting further delineation. For example, fasting tolerance is much shorter in young infants because of their larger ratio of brain versus body mass and therefore are more vulnerable to becoming hypoglycemic, when fasted for longer than 12 hours, especially if normal feeding has been impaired by anorexia, vomiting, or diarrhea because of an intercurrent illness. On the other hand, in children previously diagnosed as having ketotic hypoglycemia, an increasing number of specific metabolic defects have been recognized, such as mild forms of glycogen storage disorders (Type IXa GSD) or of ketone utilization. Thus ketotic hypoglycemia should be considered a diagnosis of exclusion and further investigation should be considered in any child with ketotic hypoglycemia who has recurrent episodes.
Several studies have shown that ketotic hypoglycemia reflects underproduction rather than overutilization of glucose. Infusions of alanine, fructose, or glycerol produce a rise in plasma glucose concentration, without significant changes in blood lactate or pyruvate levels, indicating that the entire gluconeogenic pathway from the level of pyruvate is intact, and suggesting that a deficiency of substrate rather than a defect in gluconeogenesis is involved.
Glucagon induces a normal glycemic response in affected children during the fed state, but not at the time of hypoglycemia, indicating that glycogenolytic pathways are also intact. Plasma glycerol levels are normal in these children, in both the fed and fasted states. The metabolic response to infusion of BOHB does not differ from that of normal children. Finally, the levels of hormones that counter hypoglycemia are appropriately elevated, whereas insulin levels are appropriately low.
Children with ketotic hypoglycemia have plasma alanine concentrations that are reduced in the basal state, after an overnight fast, and fall still farther with prolonged fasting. Alanine is the major amino acid used for gluconeogenesis. Its formation and release from muscle during periods of caloric restriction is enhanced by the presence of a glucose-alanine cycle, as well as by de novo formation from other substrates, such as branched chain amino acids. Hypoalaninemia in ketotic hypoglycemia probably reflects “accelerated starvation,” rather than a specific defect in alanine metabolism. As pointed out in the original description of ketotic hypoglycemia, the children are frequently smaller than age-matched controls and often have a history of transient neonatal hypoglycemia. Thus ketotic hypoglycemia may simply reflect the reduced reserve of a small muscle mass at an age when glucose demands per unit of body weight to support brain metabolism are relatively high; children with ketotic hypoglycemia may represent the lower end of the normal range for fasting tolerance. Spontaneous remission of ketotic hypoglycemia by age 8 to 9 years might be explained by the increase in muscle bulk relative to brain size, with a resultant increase in the supply of endogenous substrate, and the relative decrease in glucose requirement per unit of body mass, with increasing age.
The diagnosis of ketotic hypoglycemia is confirmed by a supervised fast. Hypoglycemia with elevated plasma FFA, BOHB, and acetoacetate develops within 14 to 24 hours in most of these children, whereas normal children of similar age can withstand fasting without developing hypoglycemia for at least 24 hours.
Episodes of ketotic hypoglycemia can be prevented or minimized by avoidance of prolonged fasting. The overnight fast should be shortened to less than 10 to 12 hours, with a bedtime snack containing carbohydrate and prompt breakfast. When episodes have been triggered by illness, parents may test the child’s urine for ketones or, preferably, plasma BOHB. The appearance of hyperketonemia precedes the hypoglycemia by several hours and indicates a need for high-carbohydrate liquids. If these cannot be tolerated, the child should be taken to the emergency department for intravenous glucose. A letter of explanation and treatment recommendation may expedite the emergency department response.
Ketotic hypoglycemia should be considered only a diagnosis of exclusion, as episodic hypoglycemia with ketosis can occur with deficiencies of several hormones or a variety of defects of gluconeogenesis or glycogen metabolism, especially hypopituitarism and GSDs. Recurrent episodes of ketotic hypoglycemia that cannot be explained by intercurrent illness should trigger reevaluation for a possible underlying disorder that had previously been missed. Among the disorders that deserve special consideration are the milder glycogen storage disorders (e.g., Type IXa GSD caused by X-linked phosphorylase kinase deficiency, PHKA2 ); adrenal insufficiency because of inhaled or intranasal glucocorticoids; or defects in ketone utilization, such as deficiency of MCT1 encoded by SLC16A1 .
Hormone Deficiency
GH and cortisol both augment glucose production, decrease glucose utilization, and accelerate lipolysis and ketogenesis. In contrast with glucagon and epinephrine, which raise plasma glucose levels within minutes, the actions of GH and cortisol are delayed for several hours and neither has a primary role in the defense against acute insulin-induced hypoglycemia. They are important, however, in preventing hypoglycemia during prolonged fasting.
Deficiency of Growth Hormone and Hypopituitarism
Deficiency of GH is an important cause of fasting hypoglycemia, especially in infants and young children. Although less common, symptomatic and asymptomatic hypoglycemia, usually precipitated by stress, starvation, or exercise, may also occur in older children and adults with either isolated GH deficiency or multiple pituitary deficiencies.
Hypopituitarism in the newborn period can cause persistent or recurrent severe hypoglycemia, and some infants may require a glucose infusion rate comparable to that required to treat congenital HI, in addition to mimicking HI by having suppressed FFA and ketone levels, and a glycemic response to glucagon. A microphallus suggests coexistent gonadotropin deficiency. Infants with congenital hypopituitarism may have cholestatic jaundice with elevated liver enzymes consistent with neonatal hepatitis. The diagnosis of GH deficiency cannot be based on a single GH value obtained at the time of spontaneous or fasting-induced hypoglycemia. Studies have shown that a single low GH value, at the time of fasting hypoglycemia, has poor specificity for the diagnosis of GH deficiency. Confirmation of the diagnosis should be based on a firm foundation of clinical evidence, including demonstration by imaging of abnormal pituitary gland development or other midline defects.
Isolated GH deficiency in young children may cause morning hypoglycemia with hyperketonemia (“ketotic hypoglycemia”). Plasma insulin levels are appropriately suppressed and the glycemic response to glucagon during spontaneous hypoglycemia is poor. The degree of ketosis at the time of hypoglycemia may be lower than expected in the more common type of idiopathic ketotic hypoglycemia, perhaps reflecting increased insulin sensitivity and impaired lipolysis. It has been suggested that the relative hypoketonemia (i.e., impaired generation of alternative fuels) may contribute to the development of fasting hypoglycemia in young children with GH deficiency. After early infancy, slow growth and low IGF-1 levels are more reliable indicators of GH deficiency, and the definitive diagnosis of GH deficiency should be confirmed by demonstrating low GH responses to standard provocative stimuli.
Growth Hormone Resistance and Insulin-like Growth Factor-1 Deficiency
Recurrent fasting hypoglycemia is common in GH resistance because of molecular defects in the GH receptor, leading to an inability to generate IGF-1, commonly referred to as Laron dwarfism . The hypoglycemia tends to improve in adolescence, although still can occur with prolonged fasting. Similar frequencies of hypoglycemia (≥ 45%) have been reported in the two largest populations with this condition, in Israel and Ecuador. The vulnerability to fasting hypoglycemia improves with treatment with synthetic IGF-1; however, one of the insulin-like effects of IGF-1 treatment is hypoglycemia, which can occur within an hour of an injection, if the child has not eaten.
Deficiencies of Cortisol and Adrenocorticotropin
Cortisol is crucially important in supporting glucose production during fasting by increasing glycogen storage and gluconeogenesis, and for mobilizing FFA; cortisol also has an indirect role in the defense against hypoglycemia by enabling epinephrine to be synthesized normally in the adrenal medullae. Activity of phenylethanolamine N-methyl transferase, the enzyme that converts norepinephrine to epinephrine, depends on intraadrenal cortisol.
Cortisol deficiency increases sensitivity to insulin; for example, an increased frequency of hypoglycemia and diminishing insulin requirement are characteristic manifestations of Addison disease in type 1 diabetes.
Fasting hypoglycemia can occur in patients with both primary and secondary glucocorticoid deficiency and is more common in neonates, infants, and young children than in adolescents and adults, and in those with more severe cortisol deficiency. Hypoglycemia is most likely to occur after prolonged fasting or during the stress of illness, especially if the usual daily glucocorticoid replacement has been interrupted by illness.
ACTH deficiency combined with GH deficiency, especially in congenital hypopituitarism, is more likely to cause hypoglycemia than primary adrenal insufficiency. Congenital ACTH deficiency, presenting with early hypoglycemia, has been reported with mutations of several genes involved in pituitary development ( POU1F1 , PROP1 , TPIT ). ACTH insufficiency is not always clinically apparent in early infancy but may manifest later in childhood.
An increasing number of specific genetic defects resulting in isolated ACTH deficiency or resistance have been identified. Red hair and severe early-onset obesity are characteristic features of children with ACTH deficiency caused by proopiomelanocortin (POMC) mutations. Hypoglycemia in an infant caused by ACTH deficiency has been attributed to a defect of prohormone cleavage. Idiopathic ACTH deficiency can be acquired in childhood, adolescence, or later adult life. Circumstantial evidence, such as an association with autoimmune thyroiditis, suggests autoimmune hypophysitis as a common cause and in some instances antipituitary antibodies have been demonstrated. In ACTH resistance, hyperpigmentation accompanies other manifestations of adrenal insufficiency.
Iatrogenic suppression of adrenal function is a common cause of glucocorticoid insufficiency, but it may be overlooked because the typical electrolyte abnormalities of mineralocorticoid deficiency are absent. An adrenal crisis with prostration, vomiting, hypotension, and hypoglycemia can be triggered by a stressful event, in a child recently weaned from high-dose glucocorticoid therapy, and has also been reported on alternate day low-dose therapy. More often, hypoglycemia is caused by inhaled and nasal glucocorticoid preparations that are the mainstay of asthma and allergy treatment for millions of children. A serum cortisol level of less than 3 mcg/dL between 7 and 9 AM in a child receiving topical or inhaled glucocorticoids is diagnostic of adrenal insufficiency; if the level is 3 mcg/dL or higher, an ACTH stimulation test is required to evaluate adrenal function.
Hypoglycemia may be a presenting feature of primary adrenal insufficiency, and often occurs during illness or starvation in patients being treated with replacement glucocorticoids. Primary adrenal insufficiency is associated with deficient epinephrine responses to hypoglycemia, and hypoglycemia manifested by neuroglycopenic rather than adrenergic symptoms may be less recognizable. Hypoglycemia is relatively frequent in treated patients with congenital adrenal hyperplasia.
As has been noted earlier for GH deficiency, the diagnosis of cortisol or ACTH deficiency, as the cause of a child’s hypoglycemia, cannot be based on a single low cortisol value during hypoglycemia, and requires additional clinical evidence (such as a concomitant markedly elevated plasma ACTH concentration in the case of primary adrenal insufficiency) or a standard adrenal stimulation test.
Deficiency of Glucagon and Epinephrine
The importance of glucagon and epinephrine in the counterregulatory responses to hypoglycemia suggests that isolated deficiency of either hormone would be likely to cause hypoglycemia, and yet no instances of hypoglycemia caused by isolated, primary deficiency of either hormone have been well documented in children.
Although there have been case reports of reduced epinephrine excretion in patients with hypoglycemia, evidence from patients with type 1 diabetes and insulinomas suggests that reduced epinephrine excretion is likely to be the consequence rather than the cause of recurrent hypoglycemia. The defective epinephrine response that accompanies adrenal insufficiency can be at least partly restored by glucocorticoid replacement. Severe fasting hypoglycemia has been reported in a young child receiving the beta-blocker, propranolol.
Despite its crucial importance in acute glucose counterregulation, there have been no well-documented cases of childhood hypoglycemia caused by isolated glucagon deficiency. One case, often cited as an example of isolated glucagon deficiency, was subsequently proven to be an instance of familial SCHAD-HI.
Genetic Disorders of Gluconeogenesis
In the early postabsorptive period, the plasma glucose level is maintained by both glycogenolysis and gluconeogenesis. As fasting continues beyond 6 to 10 hours and liver glycogen is depleted, gluconeogenesis contributes an increasing proportion of circulating glucose (see Fig. 23.3 ). Alanine from muscle is a principal substrate for gluconeogenesis, but lactate derived from peripheral tissue glycolysis also contributes to glucose production via the Cori cycle, and in later stages of fasting glycerol, derived from lipolysis of adipose tissue triglycerides, becomes a major source of gluconeogenic substrate (FFA cannot be converted to glucose). Genetic defects of the four enzymes of gluconeogenesis required to bypass irreversible steps in glycolysis (pyruvate carboxylase, phosphoenolpyruvate carboxykinase, fructose 1,6-diphosphatase [FDPase], and G6Pase) cause hypoglycemia, characterized by marked elevations of lactate (see Fig. 23.5 ; deficiency of G6Pase is described previously under the Glycogen Storage Disorders).
Pyruvate Carboxylase Deficiency
Pyruvate carboxylase is a biotin-containing protein consisting of four subunits that converts pyruvate to oxaloacetate, as the first step in the gluconeogenic pathway. Activation is dependent on acetyl CoA and occurs principally during mobilization of FFA during fasting. Several forms of deficiency have been described, typically presenting as severe lactic acidosis and encephalopathy in early infancy, usually triggered by metabolic decompensation during illness. Hepatomegaly is common. Hypoglycemia is not invariably present in pyruvate carboxylase deficiency. Urine contains large amounts of alpha-ketoglutarate. The diagnosis can be confirmed by direct sequencing of the PC gene on 11q13. Management consists of frequent carbohydrate meals and intravenous dextrose support during illness.
Phosphoenolpyruvate Carboxykinase Deficiency
Phosphoenolpyruvate carboxykinase (PEPCK) catalyzes the conversion of oxaloacetate to phosphoenolpyruvate as the second key step in gluconeogenesis. Cytosolic and mitochondrial isoforms are encoded by PEPCK1 and PEPCK2 , respectively. Deficiencies of both forms have been reported in rare patients, but the clinical phenotype remains uncertain. It is only recently that patients with deficiency of the cytosolic form of PEPCK have been confirmed at the molecular level. These patients presented with intermittent episodes of hypoglycemia, without marked elevations of plasma lactate, but with increased urinary concentrations of tricarboxylic acid cycle intermediates. Several patients with deficiency of the mitochondrial form of PEPCK have been described based on enzymatic data, but have not been confirmed at the molecular level. These patients presented with hypotonia, hepatomegaly, failure to thrive, lactic acidosis, and hypoglycemia.
Fructose 1,6-Diphosphatase Deficiency
FDPase, or fructose 1,6-bisphosphatase, catalyzes the conversion of fructose 1,6-diphosphate to fructose-6-phosphate. FDPase is encoded by FBP1 on chromosome 9q22.32. Deficiency of FDPase impairs the formation of glucose from lactate; glycerol; gluconeogenic amino acids, such as alanine; and fructose.
Hypoglycemia caused by FDPase deficiency was first described in 1970 by Baker and Winegrad. FDPase deficiency may present with hypoglycemia and lactic acidosis in the first days of life, but up to 50% of cases present later. Typical manifestations include hyperventilation because of lactic acidosis and ketoacidosis, seizures and coma from hypoglycemia, and hepatomegaly. Attacks are precipitated in infancy and childhood by prolonged fasting during intercurrent illness. Uric acid is elevated similar to type 1 GSD, secondary to sequestration of phosphate as fructose phosphates in the liver, and increased degradation of adenine nucleotides. The diagnosis should be suspected when fasting leads to hypoglycemia and lactic acidemia, with a poor glycemic response to glucagon. The diagnosis may be confirmed by genetic mutation analysis of FBP1 . Treatment of acute attacks of hypoglycemia is with intravenous glucose and bicarbonate infusion. Chronic treatment is avoidance of prolonged fasting and a reduction, but not total elimination, of fructose from the diet.
Hereditary Fructose Intolerance
Hereditary fructose intolerance (HFI) was first described as an idiosyncrasy to ingestion of fructose, and was later determined to be caused by recessive mutations of aldolase B ( ALDOB at 9q31.1). Aldolase B plays an essential role in fructose metabolism by converting fructose-1-phosphate to dihydroxyacetone phosphate and glyceraldehyde, which are substrates for either gluconeogenesis via fructose 1,6-diphosphate or oxidation via conversion to pyruvate. Ingestion of fructose (e.g., as sucrose in fruit) in patients with aldolase B deficiency causes accumulation of fructose-1-phosphate in liver, kidney, and intestinal cells, leading to intracellular phosphate depletion and both acute symptoms and potentially long-term organ damage. Phosphate depletion induces hypoglycemia by impairing glycogenolysis and, as noted earlier in G6Pase deficiency and FDPase deficiency, also causes hyperuricemia because of increased adenine nucleotide degradation. The hypoglycemia of HFI occurs immediately after fructose ingestion, rather than with fasting.
HFI presents in infancy, after fructose is introduced to the diet in fruits or as sucrose (table sugar). Fructose ingestion induces vomiting, abdominal pain, diarrhea, and hypoglycemia. With ingestion of substantial amounts of fructose, plasma lactic and uric acid levels rise and phosphorus, potassium, and bicarbonate levels fall. The severity of symptoms is proportional to the amount of fructose ingested; in early infancy, large amounts (e.g., in milk or formula made with sucrose) may produce shock, acute liver failure, and death. Older children are less likely to ingest toxic amounts because of abdominal pain induced by fructose. Acute treatment of hypoglycemia by intravenous glucose rapidly reverses symptoms and affected patients remain healthy if fructose exposure is discontinued. Chronic fructose ingestion causes failure to thrive and increasing liver dysfunction. The earliest renal effect is proximal tubular damage with glucosuria and phosphaturia; prolonged exposure to fructose can lead to kidney failure.
Fructose challenge is potentially dangerous and is not needed to make the diagnosis, because genetic mutation testing of ALDOB is available. Chronic treatment is the avoidance of all fructose-containing foods (fruits, sucrose, high-fructose corn syrup); because sorbitol is metabolized to fructose, it needs to be excluded as well.
Defects of Fatty Acid Oxidation
Fatty acid oxidation is the major source of energy for the heart and working skeletal muscle and, apart from the brain, becomes the body’s dominant fuel during prolonged fasting. Lipolysis of adipose tissue triglyceride stores releases glycerol, as a substrate for hepatic gluconeogenesis, and FFA, which are used directly as fuel for peripheral tissues and, indirectly, to spare glucose consumption. FFA are converted by mitochondrial ß-oxidation in liver to ketones (BOHB and acetoacetate), which are an alternative fuel for the brain. The key steps in transport of fatty acids into mitochondria, the ß-oxidation cycle, and the ketone synthesis pathway are outlined in Chapter 7 . Genetic disorders in all of these steps have been identified which, in general, present with life-threatening attacks of hypoketotic hypoglycemia, coma, and cardiovascular collapse induced by fasting stress. Defects interfering with early steps in long chain fatty acid oxidation tend to have more systemic features, including chronic cardiomyopathy and skeletal muscle weakness, whereas defects in later steps affecting shorter chain fatty acid oxidation, such as medium chain acyl-CoA dehydrogenase (MCAD) deficiency, may present chiefly with acute episodes of illness triggered by fasting. As a group, genetic disorders of fatty acid oxidation are relatively common (the incidence of MCAD deficiency is ~ 1 in 15,000 births in the United States, and 1 in 5000 in Northern Germany), and must be considered in the differential diagnosis of any child with hypoglycemia. Newborn screening programs exist in most countries, which can identify most of the genetic defects in fatty acid oxidation, based on analysis of acylcarnitine profiles (exceptions include HMG-CoA synthase deficiency). However, pediatric endocrinologists must also be able to recognize these fatty acid oxidation disorders, because many cases may present as new onset hypoglycemia at ages ranging from the newborn period through infancy, and up to several years of age.
Hypoglycemia can manifest in the first days of life (see Chapter 7 ) in babies exposed to early fasting stress because of failed breastfeeding. However, typically, patients may not present until several months of age, when feeding intervals overnight lengthen to 8 to 12 hours of fasting. Genetic disorders of fatty acid oxidation are especially likely to become clinically apparent during the accelerated starvation of gastrointestinal illness. Because fatty acid oxidation disorders impair ketogenesis, hypoglycemia associated with these disorders is hypoketonemic; however, in contrast to HI disorders, plasma concentrations of FFA are increased. Acute attacks of illness may be accompanied by coma, fatty liver, and encephalopathy, which mimic Reye syndrome, and may not immediately respond to glucose. Episodes may be rapidly fatal and can resemble sudden infant death syndrome. Hypertrophic or dilated cardiomyopathy and skeletal muscle weakness are prominent features of certain defects, such as long chain acyl CoA dehydrogenase deficiency, carnitine transport deficiency, and carnitine-palmitoyl transferase 2 deficiencies. In patients with primary carnitine deficiency caused by the plasma membrane carnitine transporter ( SLC22A5 ), treatment with high-dose carnitine supplementation is required; the value of carnitine supplementation in other disorders is controversial. Diagnosis can be based on an abnormal plasma acylcarnitine profile and confirmed by genetic mutation analysis.
The primary treatment of these disorders of fatty acid oxidation is avoidance of fasting. In younger children, fasting should be limited to less than 8 to 10 hours; older children may tolerate fasting as long as 10 to 12 hours. Episodes of acute illness with fasting should be treated by rapid institution of intravenous dextrose to raise plasma glucose to the high normal range.
Hypoglycemia Induced by Exogenous Agents
In children, hypoglycemia caused by exogenous agents usually represents accidental ingestions. Ethanol, salicylates, quinine, and beta-blockers, such as propranolol, are the agents most frequently implicated in childhood hypoglycemia.
Alcohol-Induced Hypoglycemia
Ethanol is a notorious cause of severe fasting hypoglycemia in young children and malnourished alcoholic adults, and occasionally in healthy adolescents who are intoxicated. Oxidation of ethanol in the liver causes an increase in the nicotinamide adenine dinucleotide (NAD)H:NAD + ratio that impairs conversion of lactate to pyruvate and, thus, inhibits gluconeogenesis. This inhibition of gluconeogenesis can lead to hypoglycemia in the setting of fasting for long enough to deplete liver glycogen stores. Young children fasted overnight are particularly vulnerable, as are adolescents who have become intoxicated with alcohol without eating. The hypoglycemia is associated with lactic acidemia and hyperketonemia. Treatment with intravenous dextrose is required to reverse the hypoglycemia.
Salicylate Intoxication
In high doses, aspirin and other salicylates have been reported to cause hypoglycemia in both children and adults. Suggested mechanisms include stimulation of insulin release or inhibition of hepatic gluconeogenesis. Infants appear to be more susceptible than older children to salicylate-induced hypoglycemia. Because of concerns about a possible role of salicylates in causing Reye syndrome, their routine use in young children has been largely abandoned.
Hypoglycin A and Jamaican Vomiting Sickness
Hypoglycemia caused by ingestion of unripe ackee fruit was first described as Jamaican vomiting sickness occurring in young children who were seeking potential foods because of undernutrition/starvation. The responsible toxin was identified as hypoglycin A, which is metabolized to a tricyclic compound that is a suicide-substrate for medium chain acyl-CoA dehydrogenase. A similar illness has been recently linked to ingestion of unripe litchi fruit, which contains a related toxin (methylene cyclopropyl-glycine). The toxins in both fruits produce a condition similar to MCAD deficiency (described earlier and in Chapter 7 ).
Hypoglycemia in Fasting, Starvation, Illness, and Stress
During prolonged fasting, FFA and ketones supply a progressively greater proportion of fuel and plasma glucose levels may fall below 50 mg/dL (2.8 mM), without autonomic or neuroglycopenic symptoms, or apparent ill effects because elevated levels of ketones provide adequate alternative fuel for the brain (see Fig. 23.2 ). This is especially likely to be seen in healthy young children and young women, because of their relatively smaller body stores of fuel and smaller body mass relative to brain size. Biochemical hypoglycemia, for example, is occasionally observed in younger children with excessive preoperative fasting.
The risk of biochemical, occasionally symptomatic, hypoglycemia is further increased in children during a wide variety of stressful illnesses, such as rewarming after profound hypothermia and near-drowning.
Hypoglycemia During Starvation and Malnutrition
Chronic malnutrition compromises the ability to maintain euglycemia, and hypoglycemia in malnourished infants and adults can be difficult to reverse. In African children with kwashiorkor, hypoglycemia has been reported to be a common terminal event. In the developed world, anorexia nervosa is an important cause of severe malnutrition, and hypoglycemia is common and may occasionally be severe.
Hypoglycemia With Prolonged Exercise
Muscle uses glucose at a high rate during exercise; however hypoglycemia is normally prevented during prolonged exercise by a decrease in insulin secretion and increased secretion of glucagon and catecholamines. Nevertheless, hypoglycemia occasionally occurs during intense exercise, even in presumably healthy adults, and can be prevented by ingestion of complex carbohydrates, but not extra sugar. Cases of hypoglycemia severe enough to cause seizures after a marathon or comparable endurance exercise have been reported. Exercise-induced HI because of promoter mutations of MCT1 (described previously) may explain at least some cases of hypoglycemia associated with exercise.
Diarrheal Illness
Hypoglycemia is uncommon in previously healthy children with acute viral gastroenteritis, unless there has been a period of starvation (as occurs after prolonged consumption of water or sugar-free fluids) or of severe diarrhea. Hypoglycemia in these settings is typically ketotic and has a good outcome. Because hypoglycemia is uncommon in otherwise healthy children, and previously unsuspected metabolic or hormonal disorders may present to an emergency department with hypoglycemia, it is important that a history of prolonged starvation be confirmed and that underlying conditions, especially the disorders of fatty acid oxidation, be ruled out.
When diarrhea (e.g., cholera or shigella) develops in severely malnourished children, hypoglycemia is a grave prognostic sign and may be fatal because of depletion of gluconeogenic substrates and alternative fuels.
Sepsis
Infectious diseases rarely cause hypoglycemia; indeed, hyperglycemia because of adrenergic stress responses is much more likely. However, septicemia, especially meningococcemia (unrelated to adrenal insufficiency), may present with severe hypoglycemia. Research in animals suggests that cytokines, such as tumor necrosis factor may amplify glucose uptake. Limited data in humans indicate appropriate suppression of insulin and elevation of counterregulatory hormones, as well as cytokines.
Hypoglycemia in Specific Infections
Hypoglycemia has been reported to occur in approximately one-third of children with severe malaria and is a risk factor for increased mortality. Insulin levels are appropriately low, whereas lactate and alanine levels are high, suggesting that hepatic gluconeogenesis is impaired. Management should include provision of glucose and monitoring of plasma glucose concentrations. In addition, drugs for malaria treatment (in particular quinine) may aggravate hypoglycemia because of their ability to stimulate insulin release.
Hypoglycemia has been repeatedly reported in infants with pertussis infection, which is caused by hyperinsulinemia and is not simply caused by fasting. Hyperinsulinemia, but without hypoglycemia, has also been shown to occur in mice after pertussis immunization. Evidence suggests that pertussis toxin amplifies the insulinogenic response to glucose rather than stimulating insulin secretion per se.
Hypoglycemia in Organ System Disease
Hypoglycemia is common in critically ill patients of all ages and can occur in failure or severe disease of nearly every major organ system. The liver is the principal source of glucose during the postabsorptive period, and under experimental conditions, hypoglycemia occurs after loss of 80% of the liver. Hypoglycemia in human liver disease is less predictable. In cirrhosis and progressive liver failure, glucose levels usually remain normal, even in patients with hepatic coma. However, fasting hypoglycemia can occur sporadically in many forms of liver disease, most likely caused by impaired glycogen reserves, with little dependence on the severity of liver impairment by other measures. Hypoglycemia has been reported in adults and children, with drug-induced hepatic injury, poisoning, and in patients with infectious hepatitis and other causes of hepatic injury. Hypoglycemia can also occur as a complication of various rare genetic diseases affecting the liver. For example, citrin deficiency has an intermediate, childhood-onset form characterized by growth retardation, episodic neurologic and behavioral abnormalities with hyperammonemia, and sometimes hypoglycemia.
Renal gluconeogenesis also normally contributes to maintenance of blood glucose during fasting, and hypoglycemia may occur in patients with end-stage chronic renal failure. Because autonomic responses to hypoglycemia are often impaired in chronic renal failure, neuroglycopenic manifestations typically predominate. In many cases of hypoglycemia with renal disease, the hypoglycemia reflects a complication of dialysis or other procedures and the existence of a specific “uremic hypoglycemia” has been challenged.
Many cases of fasting hypoglycemia associated with both acute and chronic pancreatitis have been reported in adults and children. Both hypoglycemia and hyperglycemia are recognized complications of mumps pancreatitis in children.
The causes of hypoglycemia associated with severe heart disease, both cyanotic congenital heart disease in young children, and congestive heart failure in older children and adults are complex, with evidence of both increased glucose uptake and impaired gluconeogenesis. In infants, hypoglycemia can cause heart failure that improves with restoration of normal glucose levels.
Skeletal muscle contributes substrates to gluconeogenesis during fasting, and fasting hypoglycemia occurs with several forms of muscular dystrophy and spinal muscle atrophy characterized by reduced muscle mass. A novel mechanism for hypoglycemia, resulting from intracranial disease, would be alteration of the afferent or efferent limbs of hypothalamic glucose sensing; this has been demonstrated in rodents and has been invoked in rare cases of hypoglycemia accompanying brain tumors and trauma.
Hypoglycemia accompanied by lactic acidosis has been reported as both uncommon presentations and end-stage events in patients with acute and chronic leukemias, and lymphomas. Children on prolonged chemotherapy regimens have been found to have a high rate of fasting hypoglycemia caused by depleted glycogen and gluconeogenic precursors and to direct effects of oral purine analogs.
Hypoglycemia in the Intensive Care Unit
Hypoglycemia is common in critically ill children, is often asymptomatic and, especially when associated with increased glucose variability, is associated with poorer outcomes (increased morbidity, length of stay, and mortality rates). In addition to processes specific to the underlying diseases, other contributing factors may include substrate depletion, accelerated glucose consumption, undernutrition, impaired gluconeogenesis, cytokine effects, and adrenal insufficiency. Iatrogenic factors, such as misplaced infusion lines, changes in the rate of intravenous dextrose administration, or drug effects need to be considered. Abrupt discontinuation of parenteral nutrition may lead to hypoglycemia, particularly in children younger than 3 years of age; accordingly, a gradual taper of the infusion rate is recommended to reduce the risk of reactive hypoglycemia. Factors affecting reliability of glucose measurement (e.g., altered hematocrit, oxygenation, line draws, drugs) are more common in the intensive care unit (ICU). As noted earlier, it is important to not forget the possibility that an acute illness is unmasking a previously compensated disorder of glucose metabolism, especially in a young child.
Insulin use aiming for tight glycemic control to improve outcomes in critically ill children has recently become the most common cause of hypoglycemia in the ICU. However, a recent multicenter randomized clinical trial showed that critically ill children with hyperglycemia did not benefit from tight control, 80 to 110 mg/dL, as compared with target plasma glucose levels of 150 to 180 mg/dL.
Reactive Hypoglycemia and “Spells”
Reactive hypoglycemia refers to symptomatic hypoglycemia occurring 1 to 4 hours after a meal, and is rare in children, apart from those who have had gastric surgery (fundoplication for gastroesophageal reflux or gastric bypass surgery for obesity). However, children with symptoms attributed to hypoglycemia are not infrequently referred to endocrinology clinics. Whipple’s triad (symptoms typical of hypoglycemia, associated with a documented low plasma glucose level and relieved by treatment to raise glucose) is suggested by guidelines from the Pediatric Endocrinology Society and the Endocrine Society as necessary before entertaining a diagnosis hypoglycemia, because the symptoms of hypoglycemia are very nonspecific. Although home blood glucose meters may be helpful in testing the possibility of hypoglycemia, caution is needed in considering their use because of problems with accuracy of the results (see later in Artifactual Hypoglycemia). An OGTT should not be relied on to diagnose reactive hypoglycemia, because many normal individuals develop asymptomatic low glucose 2 to 4 hours after the glucose load. Minor dietary modifications to avoid high glycemic index foods and inclusion of foods that are more slowly digested, together with protein and fat, are usually sufficient to control symptoms in reactive hypoglycemia.
Artifactual Hypoglycemia
A low plasma glucose level discovered unexpectedly in an apparently “well” child, who had an outpatient chemistry profile for reasons unrelated to carbohydrate metabolism, is most often a spurious result. The most common cause of artifactual hypoglycemia is improper handling of samples before their arrival in the laboratory. If blood is drawn and allowed to stand, glucose is consumed by both erythrocytes and leukocytes. If a whole blood sample is allowed to clot at a room temperature, the serum glucose concentration may decrease by 7 to 20 mg/dL/h. The rate of glycolysis is temperature dependent and immediate refrigeration of samples reduces but does not completely eliminate glucose consumption. Use of collection tubes that separate cells from serum also prevents glycolysis. Fluoride inhibits but does not eliminate glycolysis, and glucose levels may decrease by 20% or more when processing is delayed for even a few hours. Postphlebotomy glycolysis consumes a relatively higher proportion of glucose when the specimen contains a low glucose level at the time it was obtained, and spurious hypoglycemia is more common when either the red cell or white cell counts are elevated, and has also been described in a hemolytic crisis with a high count of nucleated red blood cells.
In 2013 the International Organization for Standardization (ISO) stated that blood glucose meters are acceptably accurate if 95% or more of measurement results are within ± 15 mg/dL or ± 15% (whichever is larger) of reference values. Thus according to ISO, if the true blood glucose is 60 mg/dL, measured glucose could be 45 to 75 mg/dL. The 2014 US Food and Drug Administration (FDA) guide for over-the-counter blood glucose meters requires 95% or more of results to be within ± 15% of reference values and 99% or more of results within ± 20% across the whole glycemic range. Based on the FDA criteria, if the true blood glucose is 60 mg/dL, measured glucose would be considered acceptably accurate in the range of 51 to 69 mg/dL. Recent evaluations of commercially available blood glucose meters show considerable differences in performance in the low glucose range. Moreover, accuracy in the low glucose range varies considerably among meters; for example, one study showed that when the reference plasma glucose was less than 70 mg/dL, mean absolute differences of various meters ranged from 2 to 11 mg/dL and mean absolute relative differences (MARD) ranged from 4.3% to 24.2%. In another study, MARD ranged from 8.9% to 39.2%, when reference plasma glucose was less than 70 mg/dL.
Continuous glucose monitoring (CGM) devices are increasingly being used to track patterns of glucose, but absolute nadirs may be inaccurate and their usefulness in evaluating hypoglycemia is still uncertain. There are concerns about accuracy of CGM at low glucose concentrations; nonetheless, in one study using a glucose monitoring system, a majority of parents of children with congenital HI found the glucose trend to be useful. CGM has also been used to signal impending hypoglycemia in studies of tight glycemic control, in critically ill children, and although potentially useful, their use has not been systematically evaluated in the diagnosis and management of diverse hypoglycemia disorders. Several reports have suggested that use of CGM may decrease the frequency and duration of hypoglycemia in high-risk newborn infants. Trials are needed in patients with congenital HI before CGM can be recommended as a routine alternative method for monitoring glucose levels during treatment. There are also no data on the use of CGM for diagnosis of hypoglycemia, such as using it in the outpatient setting as an alternative to an inpatient fasting test.
Despite the aforementioned considerations, it is important to stress that unexpected low glucose levels should not be ignored because of the possibility that a “fluke” laboratory test may detect an unrecognized chronic hypoglycemic condition.
Emergency treatment of hypoglycemia
Once the critical sample has been obtained, a minibolus of 0.2 g/kg of dextrose should be administered by intravenous infusion over 1 minute (2 mL/kg of 10% dextrose). This should be followed by a continuous intravenous infusion of 8 mg/kg/min using dextrose 10% solution. This rate of glucose administration may be rapidly and conveniently calculated by using the simple formula that 5 mL/kg/h of a 10% dextrose solution provides approximately 8 mg/kg/min of glucose. Glucose levels should be determined 15 minutes after the bolus has been given and while the maintenance glucose infusion is running. If hypoglycemia recurs, a bolus of 0.5 g/kg may be given (5 mL/kg of dextrose 10%) and the glucose infusion increased by 25% to 50%. For details of specific treatments of individual disorders, see the sections on each condition.
Appendix: Useful Tests for Diagnosis of Hypoglycemia Disorders
Diagnostic Fasting Test : Maximum duration of fast is based on age: younger than 1 month 18 h, 1–12 months 24 h, 1–12 years 36 h, older than 13 years 72 h. Time intervals between the samples are based on the plasma glucose (PG) levels: monitor PG and plasma BOHB using respective bedside meters every 3 hours until PG is less than 70 mg/dL, then every hour until PG is less than 60 mg/dL, then every 30 minutes until PG is under 50 mg/dL. Ending criteria for fast include: maximum duration (based on patient age), plasma glucose confirmed less than 50 mg/dL, BOHB 2.0 (or more) mmol/L × 2, or severely symptomatic. After confirming that PG is under 50 mg/dL (e.g., in STAT laboratory measurement), draw the critical blood samples for laboratory tests (monitoring at intermediate time points of insulin, lactate, BOHB, FFA is often useful, especially if acute changes might occur in response to patient developing adrenergic symptoms; Table 23.5 ).
Related posts:



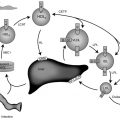
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


