The transition from intrauterine to extrauterine life requires prompt adaptations to maintain glucose homeostasis. Increased rates of glucose utilization in infants and children compared to adults heighten dependency on coordinated glycogenolysis, gluconeogenesis, and fatty acid oxidation to prevent hypoglycemia during early life. Consequently, hypoglycemic disorders often present in the neonatal period or in infancy during transitions to longer feeding intervals. Since the brain during this time is particularly susceptible to injury due to glucose deprivation, prompt recognition and treatment of hypoglycemia is paramount.
Glucose is the preferred substrate for cerebral metabolism and accounts for almost all of its energy requirements. Since the brain can neither synthesize nor efficiently store glucose, its function and health depend on a continuous glucose supply. Metabolic demands of the developing brain raise glucose utilization rates during infancy and early childhood, when the brain is a relatively larger contributor to body mass. Thus, to avoid neurological damage, diagnosing and appropriately treating hypoglycemia is particularly critical during infancy and early childhood, when adequate glucose delivery is essential for neural growth and cognitive development. To promote the prompt diagnosis and treatment of hypoglycemia by the primary care physician and endocrinologist, this chapter integrates current molecular and clinical knowledge.
Regulation of blood glucose levels in the normal state relies on a delicate balance between nutrient intake and energy expenditure. Nutrition comprises the fundamental processes by which essential nutrients are absorbed by the gut and transferred through the bloodstream and across membranes for transport into cells for metabolism. All metabolic pathways which generate adenosine triphosphate (ATP) pass through glucose or key amino acids. Thus, abnormalities in the absorption, transport, storage, and metabolism of glucose provide the mechanisms for diseases resulting in low levels of glucose in blood (hypoglycemia). Clinically, low glucose levels in key body fluids (notably blood and the cerebrospinal fluid) allow initial identification of such diseases. Beyond infancy, these disorders present rarely; thus, the astute clinician recognizes that, while many patients attribute their symptoms and problems to hypoglycemia, actual hypoglycemia is seldom documented adequately and its prevalence often overestimated.1
Adaptation to separation from energy supplied via the placenta to self-sufficiency at birth requires rapid activation of three counter-regulatory mechanisms: glycogenolysis, gluconeogenesis, and lipolysis. Thesemechanisms are immature in the newborn and even more rudimentary in premature, asphyxiated, hypoxic, or small-for-gestational age (SGA) infants. The rate of glucose turnover in newborns is 6 mg/kg/min, approximately three times the adult rate. Although the demand for glucose is high, the activities of several liver enzymes involved in glucose production (eg, glucose-6-phosphatase) and ketogenesis are diminished, particularly in preterm infants. Consequently, before the first feeding, approximately 10% of normal term newborns and approximately 67% of preterm SGA infants display blood glucose levels below 30 mg/dL in the first hours of life. These data highlight the importance of administering the initial feeding, typically the fat-rich colostrum, as soon after birth as possible.
Proper storage, oxidation, and production of glucose require intact systems for glycolysis, glycogen synthesis, glycogenolysis, and gluconeogenesis coordinated through activity of the Krebs cycle and the pentose phosphate shunt. The normal integrated regulatory effects of key hormones (eg, insulin, glucagon, and epinephrine), neural pathways, and metabolic substrates precisely match glucose utilization with the sum of exogenous glucose delivery and endogenous production. In the immediate postprandial period, exogenous glucose is absorbed in the gastrointestinal tract via sodium-dependent mechanisms, at a rate reflecting carbohydrate content and gastric emptying, and is transported into liver and pancreas via insulin-independent GLUT-2 transporters, which, unlike other GLUT transporters, are central to glucose homeostasis. In response, rising insulin levels inhibit endogenous glucose production, stimulate peripheral glucose uptake via upregulation of GLUT-4 expression in muscle and fat, and increase muscle and liver glycogenolysis. Simultaneously, inhibition of carnitine palmitoyltransferase-1 reduces peripheral free fatty acid (FFA) oxidation and hepatic β-oxidation. In this setting of increased glucose and insulin concentrations, and low FFA and ketone levels, glucose is transported into cells by GLUT-1, -3, and -4 transporters. When sensitivity to and supply of insulin is normal, glucose fluctuations before and after a meal are relatively small (eg, 60-140 mg/dL), and insulin and glucose levels return to baseline within 2 to 3 hours. Apart from periods of active carbohydrate absorption from the gut (~30 minutes after ingestion), the majority of life is spent in the fasting state. The most immediate adaptation to fasting involves release of glucose from hepatic glycogen stores, which can typically provide substrate for up to 8 hours in older children, but for much shorter periods of time in neonates and ill patients. In the fed state, glycogen only comprises about 5% of liver weight, so that in a 30-kg child energy derived from glycolysis of a total glucose supply of roughly 45 g of glucose could meet basalmetabolic demands for 5 to 6 hours. Thereafter, gluconeogenesis maintains homeostasis, and adequate 3-carbon precursors are essential as substrates for generating glucose—particularly lactate, pyruvate, alanine, glutamine, and glycerol from muscle. Both liver and kidney are capable of releasing glucose into the circulation because these tissues express key enzymes for gluconeogenesis (pyruvate carboxylase, phosphoenolpyruvate carboxykinase [PEPCK], fructose-1-6-bisphosphatase, and glucose-6-phosphatase). Gluconeogenesis is relatively impaired until late childhood as a result of the decreased availability of substrates, but, by that time, can provide 50% to 60% of glucose production after an overnight fast. Active recycling of intermediate metabolites occurs within the mitochondria of the hepatocyte and myocyte during glucose metabolism. The balance between glycolysis and gluconeogenesis appears to depend on the ambient flux of ATP in the hepatocyte, which is determined, under normal conditions, primarily by the extracellular glucose concentration. The maximal hepatic glucose output is approximately 8 mg/kg/min, which is close to the normal glucose requirement in infancy.
Liberation and utilization of fat as an energy source limits catabolism of lean body mass, while meeting energy needs in a more prolonged fasting state. Derived from adipose tissue, FFA circulates to hepatocytes for conversion to ketones: acetone, acetoacetate (AcAc; reported by most clinical laboratories unless requested otherwise), and β-hydroxybutyrate (β-OHB). Over periods of prolonged fasting, FFA oxidation accounts for almost 80% of the body’s energy sources. In contrast to gluconeogenesis, ketogenesis occurs more rapidly and to a higher level in children than in adults. After fasting for 20 hours, the ketone body turnover rate in young children is comparable to that of adults who have fasted for several days. Most body tissues (including cardiac and skeletal muscle) can use FFA for oxidative metabolism, with the notable exception of the brain, which requires β-OHB, the ketone species that predominates in human plasma (typically 10-fold over AcAc or acetone) and also readily crosses the blood-brain barrier. Ultimately, these adaptations to prolonged fasting decrease glucose turnover by approximately 50% and lead to a gradual decrease in circulating glucose concentrations.
In summary, finely tuned orchestration of glycogenolysis, gluconeogenesis, and fat oxidation normally ensures a critical arterial plasma glucose concentration which, facilitated by insulin-independent glucose transport proteins (GLUT-1 and -3), can provide a continuously adequate supply of glucose to the brain. Importantly, GLUT-1 and -3 proteins are upregulated by chronic cerebral hypoglycemia, but glucoregulatory factors, including insulin, do not affect glucose uptake into the brain.
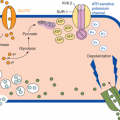
Released from β-cells in the pancreatic islets of Langerhans, insulin is the only endocrine hormone which significantly and directly lowers the circulating glucose level. It does so by mediating glucose uptake into tissues. Pancreatic β-cells and the liver integrate the major sensors responsible for detecting high glucose and stimulating insulin release. At the β-cell level of glucose-sensing, any acute rise in the intracellular concentration of ATP or free calcium induces insulin release (Figure 12-1). Glucokinase constitutes the primary sensor for high glucose within the β-cell because its enzymatic action is the rate-limiting step for glycolysis, which results in ATP production. Oxidation of many intermediate fuels, such as glutamate, results in insulin release by raisingintracellular ATP. Glutamate dehydrogenase (GDH) reversibly oxidizes glutamate to α-ketoglutarate, a direct intermediate of the Krebs cycle, for ATP production. This GDH-catalyzed reaction is enhanced by positive allosteric regulators such as L-leucine and inhibited by guanosine triphosphate (GTP).2
Figure 12-1
Mechanisms of insulin secretion in pancreatic β-cells. An increase in the ATP to ADP ratio inhibits SUR1 function, resulting in potassium channel closure, depolarization of the cell membrane, influx of calcium, and release of insulin. The ATP to ADP ratio is increased (and therefore insulin secretion stimulated) by glucose oxidation via glucokinase (GK) and by leucine stimulation of glutamate oxidation via glutamate dehydrogenase (GDH also known as GLUD1). Abnormally increased pyruvate levels in the β-cell will stimulate insulin secretion. Diazoxide keeps SUR1 open to inhibit the release of insulin. GLUT2, glucose transporter 2; MCT1, monocarboxylate transporter 1; ADP, adenosine diphosphate; ATP, adenosine triphosphate; GTP, guanine triphosphate; VDCC, voltage-dependent calcium channel; Kir6.2, inward- rectifying potassium (K-ATP) channel of SUR1/Kir6.2 complex. Dashed lines denote steps in the pathway.
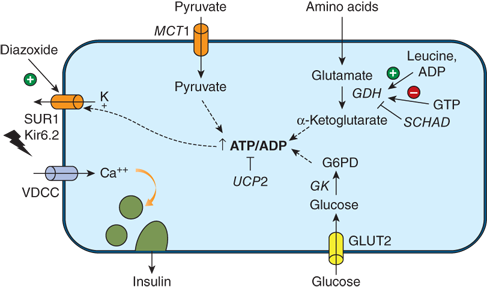
The primary pathway for the regulation of insulin secretion in humans is the sulfonylurea receptor (SUR1)-potassium channel complex, which accounts for more than 90% of glucose-stimulated insulin release. In humans, this complex consists of four subunit transporter proteins which bind ATP (ABCC8) and four inward-rectifying subunits regulating potassium (K) flux (Kir6.2 or K-ATP channel). Derived from glycolysis or other pathways, ATP binding at the intracellularnucleotide-binding site of the trans plasma membrane of SUR1 will close the associated inward-rectifying K channel. Closure to K flux depolarizes the membrane, which activates voltage-gated, L-type calcium channels. Influx of calcium raises free calcium levels intracellularly, leading to insulin release from storage granules via a pathway regulated by calmodulin-dependent kinase II.
Multiple counter-regulatory pathways have evolved to maintain normal circulating glucose concentrations (also known as euglycemia or normoglycemia) by opposing insulin action during the stress response.
In response to falling plasma glucose levels, glucagon released from pancreatic α-cells traverses the portal circulation to act primarily on 6-phosphofructo-2-kinase/fructose-2,6-bisphophatase in hepatocytes. The kinase and phosphatase portions of this bifunctional enzyme regulate, respectively, the key steps controlling glycolysis and gluconeogenesis. Glucagon favors the dephosphorylated state, resulting in activation of the kinase and inhibition of the phosphatase, enhancing gluconeogenesis while simultaneously inhibiting glycolysis.
Catecholamines acutely rise in response to hypoglycemia. Adrenergic α-receptor binding enhances amino acid uptake into the liver, which increases the availability of substrates for gluconeogenesis while β-adrenergic receptor activation stimulates lipolysis by inducing phosphorylation of hormone-sensitive lipase (the enzyme that cleaves triglycerides into FFA and glycerol within adipose tissue). There, catecholamines stimulate glycogenolysis via both the α1– and β-receptor mechanisms; in muscle, stimulation of glycogenolysis occurs solely by β2-receptor activation and requires the presence of glucocorticoids to antagonize the effects of circulating insulin. Catecholamines also enhance FFA entry and mobilize triglycerides from skeletal muscle, but inhibit the release of amino acids from skeletal muscle, preserving this resource in muscle during stress. Effects on insulin secretion are complex and incompletely understood; β2-adrenergic activation appears to stimulate insulin and glucagon secretion while α-receptor stimulation suppresses insulin secretion and stimulates glucagon secretion. Finally, catecholamines acutely inhibit insulin-mediated glucose uptake via β-adrenergic stimulation.
Growth hormone (GH) is released in response to stress, fasting, hypoglycemia, and rapid declines in blood glucose concentrations. In the neonatal period, GH provides a primary defense against hypoglycemia via its inhibition of peripheral insulin-stimulated glucose uptake, perhaps by action of specific insulin-like growth factor (IGF)-binding proteins to induce insulin resistance. GH also stimulates release of FFA from adipose tissue stores, thereby providing an alternate fuel to glucose during prolonged states of hypoglycemia.
Hypoglycemia of significant duration (eg, hours) is a more potent stimulus than is acute hypoglycemia for GH, ACTH, or cortisol release, but this may not be the case when hypoglycemia is persistent or recurrent. For that reason, a low serum GH level found at the time of spontaneous, as opposed to acute induced, hypoglycemia does not necessarily imply GH deficiency as a cause of the hypoglycemia.
Glucocorticoids, synthesized in the adrenal cortex upon ACTH stimulation, enter the adrenal medulla and stimulate the activity of phenylethanolamine-N-methyltransferase (PNMT), which catalyzes the N-methylation of norepinephrine to epinephrine. ACTH and cortisol deficiency, therefore, result in a decrease in immediate counter-regulation by epinephrine production and secretion. Furthermore, cortisol promotes gluconeogenesis via mobilization of amino acids from muscle and activation of glucose-6-phosphatase and phosphoenolpyruvate carboxykinase.
In summary, the main insulin counter-regulatory hormonal action during stress arises from adrenomedullary epinephrine, with contributions from adrenocortical cortisol, pancreatic glucagon, and pituitary GH. Although rare tumors can secrete IGFs which cross-react with insulin receptors, IGFs typically exert only permissive actions on glucose transport and metabolism. Regulation of insulin-mediated glucose uptake is the primary mechanism upon which the clinician must focus. Diseases that impact this process involve the hormones of the stress response as well as inborn errors of insulin secretion, glucose transport, and ATP metabolism.
Hypoglycemia is a common clinical problem with serious neurological sequelae when detection is delayed or treatment inadequate. Even though low blood sugar may often be transient, hypoglycemia itself is never physiological (in other words, never normal) and should not be disregarded when documented adequately. Various approaches have been applied to define an abnormally low level of circulating glucose.3 The most pragmatic relies on glucose level–dependent appearance and resolution of clinical signs upon treatment (Whipple triad, Box 12-1) (also see “Signs and Symptoms” later on in the chapter for specifics). Older epidemiological studies of at-risk groups (infants not fed promptly and frequently after birth) were troubled by ascertainment bias are not representative of a truly healthy, normal population. Physiological fasting studies, which integrate sensitive and specific endocrine assessments with safe clinical outcomes, provide the most logical and compelling information.
Box 12-1. Whipple Classic Triad3
Symptoms consistent with hypoglycemia
Low plasma glucose level (the definition based on available technology as discussed in the text)
Resolution of symptoms after raising the blood glucose level
Decades of such physiological work confirm that blood glucose at any age is normally above 70 mg/dL (3.9 mmol/L).4 While many complain that this cut-off triggers too many interventions, abundant evidence suggests that the developing brain is more susceptible to low glucose levels, which behooves clinical vigilance and use of a threshold (ie, 70 mg/dL) above which no counter-regulatory response typically is triggered to maintain glucose homeostasis. Thus, the prudent clinician feeds a newborn infant with a blood sugar less than 70 mg/dL and reassesses the blood glucose level every 15 minutes until clinical recovery is confirmed. While no specific blood glucose level will be universally accepted as the dividing line between hypo- and normoglycemia, use of 70 mg/dL as a target for normoglycemia is safe and supported by physiology.4 Three important caveats should be considered. First, this recommendation applies to the symptomatic infant with documented recurrent and/or persistent hyperinsulinemic hypoglycemia, but it may not apply to newborns with a single episode of low plasma glucose by less reliable point-of-care testing. Second, any documented plasma glucose concentration less than 60 mg/dL for infants and children should prompt thorough evaluation. Third, for reliable diagnostic information from the “critical blood sample,” a simultaneous plasma glucose £ 60 mg/dL (and preferably less than or equal to 50 mg/dL) is recommended.
Casual measurement of blood glucose by finger-stick is common clinically. Blood glucose should be assessed with any new-onset seizure disorder (preferably at the time of the convulsion) at any age, or when the patient demonstrates supraphysiological glucose utilization to maintain blood glucose greater than 70 mg/dL (ie, an intravenous glucose infusion rate > 5 to 8 mg/kg/min in infants or > 3 mg/kg/min in adolescents and adults). It is also important to assess blood glucose levels in high-risk infants, including those born SGA, premature, or large-for-gestational age (given their high incidence of known or unknown diabetic mothers).
Although portable glucose meters are convenient and relatively inexpensive, their advantages are offset by their inaccuracy (up to 20% off the true laboratory glucose reading) and unreliability (simultaneous results often vary by 10%). Point-of-care glucometers generally provide interpretable readings between 70 and 500 mg/dL. Glucometer measurements at either extreme raise concern and require further assessment and intervention. Definitive diagnosis of hypoglycemia requires proper specimen collection and prompt handling. The ideal specimen is whole blood drawn from a free-flowing intravascular catheter into a sodium fluoride (NaF)-containing, gray-top tube, which is then processed immediately by the hospital laboratory. (NaF inhibits glycolysis by erythrocytes which, prior to centrifugation, can decrease the glucose level by as much 20 mg/dL/h.) Plasma glucose results obtained promptly by a glucose oxidase assay are typically accurate and reliable within ±1 mg/dL.
Hypoglycemia results from one or more of the following:
Excessive glucose utilization owing to excess insulin or insulin-like action
Deficient energy production owing to defective glycogenolysis, gluconeogenesis, ketogenesis, or ketone utilization
Energy needs which exceed carbohydrate reserves (eg, illness with prolonged vomiting and anorexia, SGA infant)
Exogenous medications or toxic ingestions
5 Reactive hypoglycemia
A detailed list of etiologies of childhood hypoglycemia is presented in Table 12-1.
|
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


