Introduction
This chapter reviews two separate and partially overlapping types of anemia: hypochromic anemias, resulting from a decreased amount of hemoglobin in red blood cells, and hemolytic anemias, resulting from increased red blood cell destruction. Of the hypochromic anemias, iron deficiency is the most common cause, and indeed the most common overall cause of anemia worldwide. As such, concurrent iron deficiency anemia may complicate interpretation of findings and the ability to diagnose other acquired and congenital causes of anemia. Hemolysis may result from congenital or acquired defects in the red blood cells themselves or extrinsic factors resulting in increased red blood cell destruction. Although most hemolytic anemias are normochromic, the combination of hypochromia and increased hemolysis defines a distinct subset of entities, of which thalassemias are the most common. This chapter focuses on the diagnostic approach to these disorders; the broad category of macrocytic anemias is considered elsewhere.
Hypochromic Anemias
Anemia is defined in public health terms by hemoglobin thresholds and by the World Health Organization (WHO) as values in the fifth percentile of the hemoglobin concentration of the same sex and age group at the same altitude. Pregnant women are considered separately. Specific hemoglobin cut-off values for a United States population drawn from the National Health and Nutrition Examination Survey (NHANES III, 1988–1994) range with age and sex, with normal hemoglobin levels ranging from 12.2 g/dL at age 1 to 2 years to 13.1 g/dL at 6 to 11 years (mean, sea level). After age 12, male and female values differ. Mean values for women are 13.4 to 13.5 g/dL in childbearing years, 13.7 g/dL aged 50 to 69 years, and 13.6 g/dL aged 70 years and older. In men, mean values are 14.2 g/dL aged 12 to 15, 15.3 g/dL aged 16 to 49 years, 15.0 g/dL aged 50 to 69 years, and 14.5 g/dL aged 70 years or older. Reduced hemoglobin concentration may result from a decrease in the number of red blood cells as assessed by the red blood cell count (RBC) owing to increased destruction or reduced production and/or a decrease in the hemoglobin content of individual red blood cells (reported as mean corpuscular hemoglobin [MCH]). Depending on cell size, red blood cells may appear hypochromic with an increased area of central pallor (more than one-third the size of the red blood cell) and a reduced mean corpuscular hemoglobin concentration (MCHC). Table 3.1 shows a range of potential diagnostic considerations for the presence of hypochromic red blood cells. The possibility of iron deficiency anemia should be considered first because it is the most common cause and may coexist with other sources of anemia, and potentially complicate that evaluation. This is particularly true in the case of patients with previously undiagnosed thalassemia.
| Red Blood Cell Features | Hemolysis | Other | |
|---|---|---|---|
| Iron deficiency anemia | Hypochromic, microcytic, anisopoikilocytosis, pencil cells | No | RBC often reduced High RDW |
| Iron-resistant iron deficiency anemia | Hypochromic, microcytic | No | Refractory to iron supplementation; may respond to parenteral iron, elevated hepcidin |
| Vitamin A deficiency | Hypochromic, microcytic; may have increased anisocytosis, poikilocytosis | No | Increased iron stores in liver and marrow, serum levels low |
| Anemia of chronic disease | Often normocytic, normochromic, in long-standing form can be hypochromic, microcytic | Possible mild hemolysis owing to relative erythropoietin deficiency and/or cytokine-induced macrophage activation | Underlying infection/inflammation/ malignancy |
| Thalassemias | Hypochromic, microcytic, target cells, coarse basophilic stippling, polychromasia | Yes | RBC usually normal or increased; microcytosis may occur without anemia |
| Certain hemoglobinopathies (Lepore variant, HbE ) | Hypochromic, microcytic | Yes | Usually no stippling or polychromasia |
| Chronic liver disease | Macrocytic or microcytic, target cells | No | Normal or increased serum ferritin; may be complicated by iron deficiency |
| Chronic renal disease | Generally normochromic, normocytic; in some cases schistocytes, hypochromic, microcytic | Can be significant in setting of uremia, dialysis | Variable by etiology, dialysis, concurrent iron deficiency, decreased erythropoietin, decreased clearance of hepcidin |
Iron Deficiency Anemia
Iron deficiency is defined as a state in which there is insufficient iron to maintain normal physiologic function; only when it has reached a certain duration or severity does it manifest with red blood cell abnormalities. Iron deficiency may result when storage iron has been depleted, when the need for iron is increased, and when iron need exceeds intake. This occurs in physiologic circumstances, such as pregnancy and rapid growth in infancy, as well as in certain disease states.
Iron deficiency anemia affects approximately 3% of children in the United States with an additional 8% to 10% of children having iron deficiency without evidence of anemia. The problem globally is more substantial owing to dietary practices and lack of sufficient access to nutrients, potentially affecting half of children worldwide. Iron deficiency anemia is also common in women of child-bearing age, with menstrual losses and pregnancy as the most common causes. It also affects 2% to 5% of adult men and postmenopausal women in the developed world. Unexplained iron deficiency should be investigated for a potential source of bleeding and/or serious underlying pathology, such as colon or other gastrointestinal cancer, celiac disease, bleeding varices, gastric ulcer, or urologic disease. Patients taking nonsteroidal antiinflammatory agents or those who have a history of weight loss surgery may also be at increased risk.
Clinical features of iron deficiency anemia include symptoms attributable to any anemia, such as easy fatigability, shortness of breath, palpitations, headache, light-headedness, and diminished work performance. Physical findings may include pallor, bounding pulse, and systolic murmurs. Lack of sufficient tissue iron may also result in paresthesias, restless leg syndrome, angular stomatitis, glossitis, esophageal webs, abnormal food cravings (pica), koilonychia, and other findings. A combination of dysphagia, esophageal webs, and iron deficiency has classically been described as Plummer-Vinson syndrome, but this presentation is exceedingly rare.
Depletion of iron stores alone is not sufficient to cause disease, with the presentation of anemia depending on intake, physiologic demand, and duration of the deficiency. Fig. 3.1 shows a characteristic peripheral blood smear from relatively severe iron deficiency. However, several stages of iron deficiency have been described, with early stages more difficult to recognize morphologically : (1) negative iron balance, (2) fall in iron stores with a fall in serum ferritin, (3) fall in serum iron and transferrin saturation, (4) hypochromic reticulocytes, (5) reduced hemoglobin, and (6) reduced mean corpuscular volume (MCV).
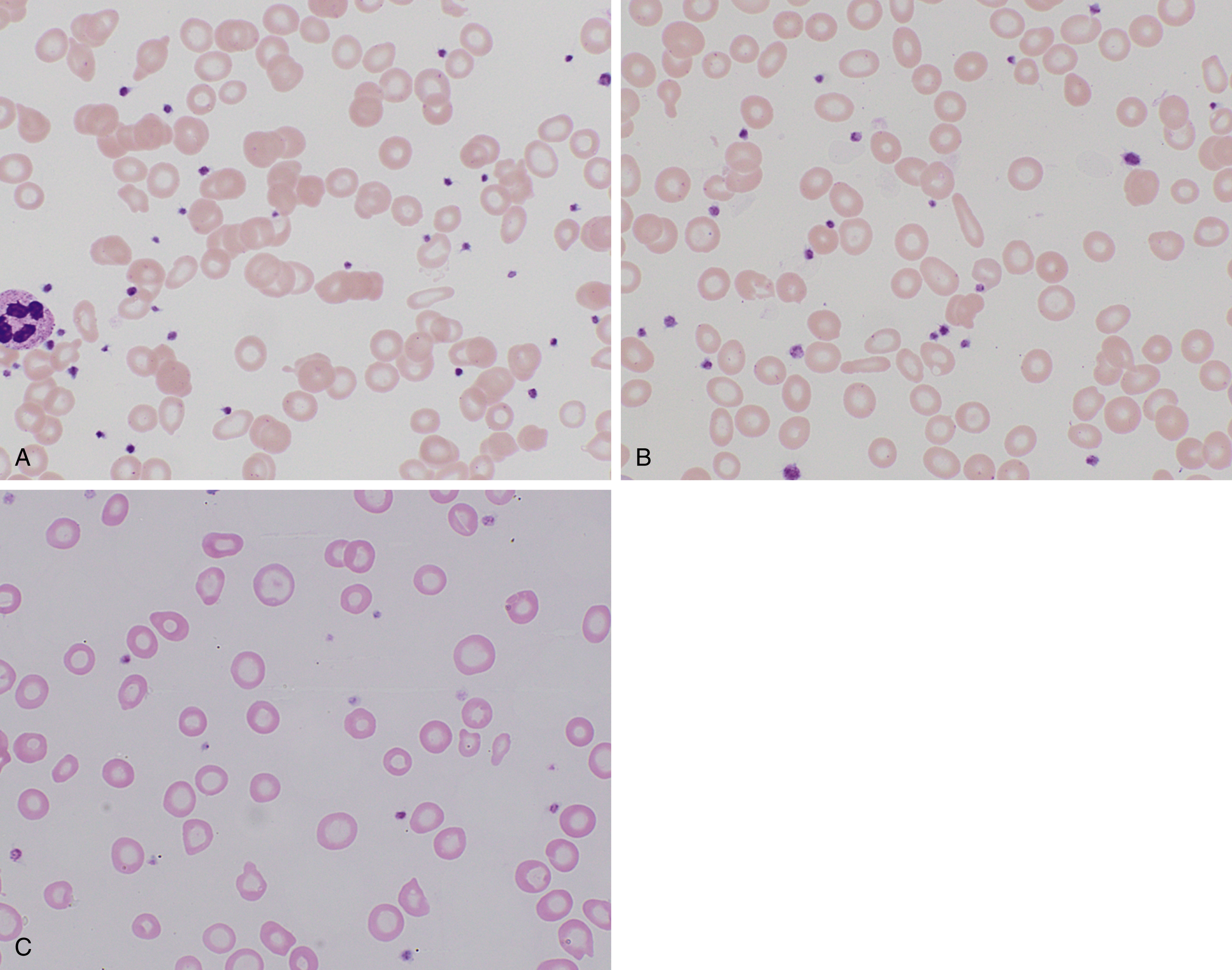
In early stages, red blood cells may be normocytic and normochromic, but the most classic presentation is that of microcytosis, marked anisocytosis (increased red blood cell distribution width [RDW]), and variable hypochromia. There is poikilocytosis with elliptical and elongated, so-called “pencil” cells as the classic finding (see Fig. 3.1 ). The poikilocytosis is thought to be secondary to increased membrane stiffness. The absolute reticulocyte count is reduced, except following iron therapy. The bone marrow may show erythroid hyperplasia in early disease but not at late stages of the disease. Erythroblasts may appear hypochromic with frayed edges ( Fig. 3.2 ). Sideroblasts are decreased, with the percentage similar to the saturation of transferrin.
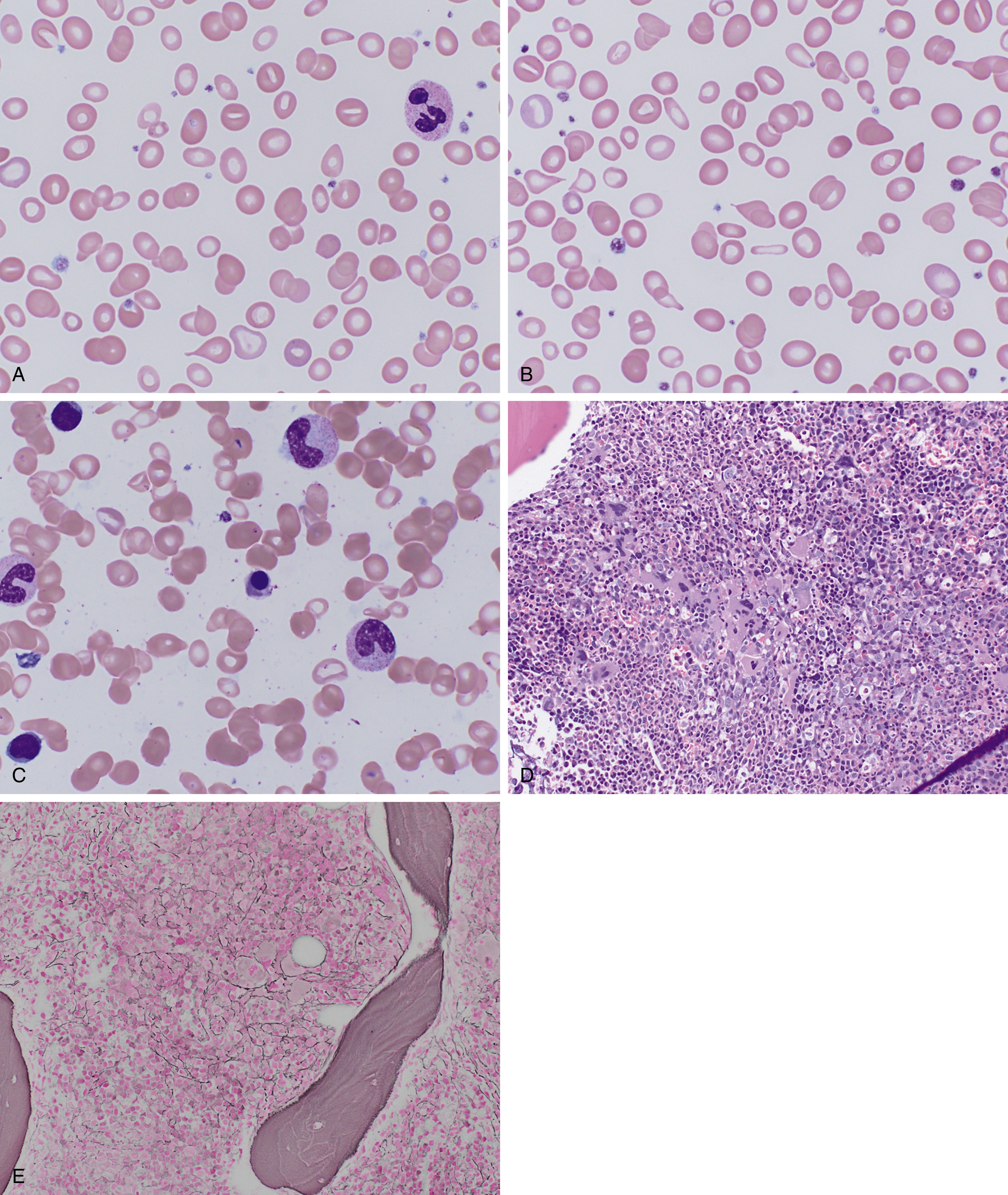
In terms of laboratory diagnosis, the tests for iron deficiency most commonly used are the serum iron, transferrin, serum ferritin, and soluble transferrin receptor (sTfR) levels. Transferrin is the major iron transport protein in the body, with levels generally reflective of nutritional status, but over time, levels are related inversely to the total body iron stores. As such, transferrin is used as a measure of the total iron-binding capacity (TIBC), and the percent saturated (as calculated from serum iron) is used in the assessment of iron deficiency. Serum ferritin generally correlates with tissue ferritin levels and the amount of storage iron, but it is also an acute-phase reactant disproportionately elevated in the setting of inflammation. Nonetheless, it remains the best laboratory measure of total body iron stores, and a low serum ferritin (<12 to 15 ng/mL) is diagnostic of iron deficiency. When in question, a bone marrow evaluation can be performed to assess iron stores directly.
An alternative method is to assess sTfR levels, which are elevated in the setting of iron deficiency anemia but low in anemia of chronic disease. Transferrin receptors are expressed on cells requiring iron, and thus erythroid tissue accounts for a large percentage of TfR expression in the body (80%). Recycling and shedding of this protein from the membrane results in circulation of sTfR and can be used to estimate the size of the erythroid pool in the marrow. Increased shedding occurs in the setting of iron deficiency, although this method does not appear superior to ferritin in detecting iron deficiency, particularly in the setting of nonhematologic malignancy. In addition, noncomplexed protoporphyrins from failed production of hemoglobin or protoporphyrins complexed with zinc instead of iron may also be of value in identifying iron deficiency.
The reticulocyte hemoglobin content has been of increasing interest because it provides an indirect measure of the functional iron available for new red blood cell production over the previous 3 to 4 days. This can provide an early measure to the response to iron therapy and help confirm the original diagnosis. Although anemia of chronic disease (see subsequent section) is also associated with reduced reticulocyte hemoglobin and generally does not respond to oral iron therapy, the identification of a functional iron deficiency may still be of value to help identify patients who will respond to intravenous iron therapy. A comparison of laboratory values for iron deficiency anemia with anemia of chronic disease is shown in Table 3.2 .
| Iron Deficiency | Anemia of Chronic Disease | |
|---|---|---|
| Serum iron | Decreased | Decreased |
| Serum TIBC, serum transferrin | Increased | Normal or decreased |
| % Saturation of transferrin | Decreased (15% to 20%) | Decreased |
| Serum ferritin | Decreased | Increased or normal |
| Erythrocyte ferritin | Decreased | Decreased |
| Serum transferrin receptors | Increased (likely secondary to increased erythroid membrane synthesis) | Normal |
| Serum hepcidin | Decreased, unless affected by comorbidities | Increased |
| Erythrocyte protoporphyrins/zinc protoporphyrin | Increased (also seen with lead poisoning) | Increased |
| Reticulocyte count a | Decreased (should respond to iron therapy) | Decreased |
| Reticulocyte hemoglobin content | Decreased | Decreased |
| Anisocytosis (RDW) | Increased | Slightly increased |
| Poikilocytosis | Increased (elliptocytes, pencil cells) | Slightly increased |
| Hemoglobin and hematocrit | Reduced more than the RBC | Mild to moderate reduction |
| Bone marrow | Early disease with erythroid hyperplasia; late at basal level; small normoblasts with frayed margins; storage iron absent | Generally normocellular; normoblasts may have frayed cytoplasm; storage iron normal or increased |
a Reticulocyte count must be corrected for anemia (patient Hct)/(normal Hct of 0.45) and correcting for the maturation time based on the level of anemia as reticulocytes are released earlier from the marrow. For hematocrits of 45%, 35%, 25%, and 15%, the reticulocyte maturation times are 1.0, 1.5, 2.0, and 2.5, respectively. Once corrected, the reticulocyte value can be compared with the normal value of 1%.
Anemia of Chronic Disease
Anemia may also result when transport of iron to target tissues is impaired by infectious, inflammatory, or malignant processes, but it does not respond to increased iron intake. So-called anemia of chronic disease or anemia of chronic inflammation is characterized by reduced red blood cell production and reduced serum iron in the setting of adequate iron stores. This disorder is more frequent in adults compared with children and may affect up to 50% of hospitalized patients. Worldwide, infections remain major sources of anemia and may significantly affect both the adult and pediatric populations. The cause of anemia in these settings may be multifactorial, but an underlying inflammatory process can be a significant component. For example, studies of serum ferritin and soluble transferrin receptor levels suggest Schistosoma mansoni infection in children that is most likely associated with an underlying inflammation rather than iron deficiency, although iron deficiency may also be a component caused by injury by the parasite to the bowel and bladder walls during egg transit.
Anemia of chronic disease is associated with increased systemic cytokines leading to elevated hepcidin, a protein produced by the liver that causes proteolysis of ferroportin. Ferroportin is the key transporter that enables cells to export iron. As such, the proteolysis of ferroportin leads to iron becoming trapped in macrophages and enterocytes with a drop in serum iron levels. Red blood cell production is reduced owing to limited iron availability. Interleukin (IL)-6 has been found to be a particularly potent inducer of hepcidin. Although in the chronic setting this process may significantly increase the morbidity of the underlying disease process, it is thought to be a protective mechanism in the setting of infection by decreasing host iron supply to invading microbes.
Elevated cytokines may result in additional adverse effects on red blood cell survival, metabolism, erythropoietin (EPO) secretion, and inhibition of bone marrow hematopoiesis. Different inflammatory and infectious processes may be associated with different cytokine profiles, resulting in variability in the presentation and severity of the disease. EPO levels may be of interest, which, although typically above normal, may be lower than expected for the degree of anemia in inflammatory conditions, resulting in hemolysis of young red blood cells, so-called neocytolysis. This may be a particular issue in chronic kidney disease. In addition to lower levels of EPO, certain cytokines may also reduce the ability of erythroid progenitors to respond to a given level of EPO, necessitating alternative dosing of EPO in patients with ongoing inflammatory or certain neoplastic processes or combined approaches to treatment. Cytokines may also exacerbate anemia through increased macrophage activation and hemophagocytosis.
In terms of diagnosis, the decrease in hemoglobin is typically limited with mild to moderate anemia. It demonstrates normocytic, normochromic red blood cell morphology, but up to 50% of patients may have microcytic, hypochromic anemia. In contrast to iron deficiency anemia, anisocytosis and poikilocytosis are slight. Laboratory values show low serum iron, normal to decreased TIBC, and decreased saturation of transferrin, but in contrast to iron deficiency they show increased serum ferritin and hepcidin levels (see Table 3.2 ).
Iron-Refractory Iron Deficiency Anemia
Rare cases of iron-refractory iron deficiency anemia may be caused by a mutation in the TMPRSS6 gene required to downregulate hepcidin and thus poorly absorb dietary iron. This results in a microcytic, hypochromic anemia with low transferrin saturation and normal/high serum hepcidin values. By contrast, patients with iron deficiency show low/undetectable hepcidin values.
Other Vitamin Deficiencies
Other vitamin deficiencies ( Table 3.3 ) may also result in hypochromic anemia with decreased red blood cell size (MCV) and MCHC. Of particular consideration in the developing world is vitamin A deficiency, estimated to affect 253 million preschool children worldwide and to co-occur frequently with and potentially exacerbate iron deficiency anemia. Vitamin A may play multiple roles in reducing anemia, including increased mobilization of iron stores from the liver, enhanced erythropoiesis, and reduced risk of infection. ,
| Vitamin/Mineral | Associated Clinical Features | Diagnostic Tests | Actions Related to Red Blood Cell Metabolism/Physiology |
|---|---|---|---|
| Vitamin A | Xerophthalmia, night blindness, Bitot spots, keratomalacia, impaired immunity | Decreased serum retinol (Ref. range: 1–3 mmol/L) Therapeutic trial of vitamin A | Growth and differentiation of erythrocyte progenitor cells, mobilization of iron stores from tissues and prevention of anemia or infection by boosting the immunity , |
| Vitamin B 2 /riboflavin | Sore throat, hyperemia of pharyngeal mucous membranes, cheilitis, stomatitis, glossitis, and seborrheic dermatitis Increased risk of cardiovascular events, esophageal carcinoma, and cervical carcinoma | Plasma riboflavin concentration (Ref. range: 1–19 μg/L) EGR assay (activity coefficient: >1.4 indicates riboflavin insufficiency) | Flavoproteins are catalysts in a number of mitochondrial oxidative and reductive reactions and function as electron transporters; may affect iron absorption |
| Vitamin B 6 /pyridoxine | Stomatitis, glossitis, cheilosis, irritability, confusion, depression, and possibly peripheral neuropathy Severe deficiency is associated with seborrheic dermatitis, microcytic anemia, and seizures | Mean plasma PLP concentration (>30 nmol/L [>7.4 ng/mL]) is sufficient level Erythrocyte transaminase activity (a functional test of pyridoxine status) Urinary 4-pyridoxic acid excretion (>3.0 mmol/day, used as an indicator of adequate short-term status) | Pyridoxal phosphate is involved in decarboxylation of amino acids involved in heme synthesis |
| Vitamin B 12 /cobalamin a | Megaloblastic anemia; skin pallor, jaundice, atrophic or acute glossitis, gastrointestinal symptoms, central nervous system degeneration (typically posterior and lateral columns of the spinal cord) | Serum cobalamin assay (200–900 ng/L) | Synthesized by microorganisms and found in nearly all animal tissues; deficiency inhibits purine and thymidylate syntheses, impairs DNA synthesis, and causes erythroblast apoptosis |
| Vitamin C/ascorbic acid | Severe deficiency leads to symptoms of scurvy, gum disease, impaired wound healing, mood disorder | Plasma levels can be measured by high-performance liquid chromatography, normal range 0.6–2.0 mg/dL; fasting more informative | Vitamin C may improve iron absorption , |
| Copper | Fragile, abnormally formed hair, depigmentation of the skin, myeloneuropathy, neurologic abnormalities, hepatosplenomegaly, osteoporosis, sideroblastic anemia, neutropenia | Low serum copper (Ref range: 0.75–1.45 μg/mL) Low erythrocyte copper-zinc superoxide levels Diminished 24-hour urinary copper excretion | Ceruloplasmin acts as a plasma ferroxidase, converting iron to a valence that can be bound by plasma transferrin |
| Folate a | Megaloblastic anemia; adequate folate reduces risk of neural tube defects | Serum and red blood cell folate | Similar to cobalamin; required for purine and thymidylate synthesis and erythroblast proliferation |
| Zinc | Growth retardation, loss of appetite, impaired immune function, hair loss, diarrhea, delayed sexual maturation, impotence, hypogonadism in males, eye and skin lesions, sideroblastic anemia | Serum and plasma zinc levels are commonly used tests (<60 μg/dL or <70 μg/dL is abnormal in nonpregnant adults) | Approximately 250 proteins contain zinc; zinc has important roles, both in cell division and apoptosis |
a Although this chapter focuses on microcytic anemias, vitamin B 12 and folate were included for the sake of completeness.
Vitamin B 6 deficiency, which functions as a coenzyme in decarboxylation and transamination of amino acids and synthesis of porphyrin precursor aminolevulinic acid, may result in hypochromic, microcytic anemia. This is rare, but can be seen in the setting of malnourishment or treatment with isoniazid or other antituberculosis agents, which interfere with vitamin B 6 metabolism. However, vitamin B 6 supplementation is generally given with isoniazid to prevent other complications, including peripheral neuropathy.
Copper deficiency may be associated with a range of anemic presentations: macrocytic, microcytic, or normocytic. Microcytic anemia is more commonly seen in the setting of ingestion of chronic, massive quantities of zinc, such as that contained in dental fixatives, resulting in secondary copper deficiency. Copper deficiency from malnourishment or following bariatric surgery is more often macrocytic. Patients may also present with neutropenia and, rarely, thrombocytopenia. Diagnosis is based on a low serum ceruloplasmin level or a low serum copper level with or without elevated serum zinc concentrations. On bone marrow evaluation, myelodysplastic features, including vacuolated myeloid and erythroid precursors and ring sideroblasts, can be appreciated, but these dysplastic features resolve following correction of zinc toxicity.
Hereditary Sideroblastic Anemia
Hereditary sideroblastic anemias, which are rare and diverse, result from a number of genetic mechanisms (X-linked, autosomal dominant, autosomal recessive) that cause pathologic deposition of iron in the mitochondria of erythroid precursors. This manifests as increased ring sideroblasts in the bone marrow and Pappenheimer bodies on peripheral smear (see Fig. 3.3 ). They can be categorized by abnormal mitochondrial heme synthesis, iron-sulfur cluster biogenesis, or abnormal mitochondrial protein synthesis. Depending on the underlying defect, this may result in decreased hemoglobin synthesis and hypochromia and/or ineffective red blood cell production. X-linked inheritance is most common (40% of cases) with decreased expression of γ-aminolevulinic acid synthase, most commonly owing to a point mutation in the ALAS-2 enzyme. Deletions in the mitochondrial genome may also result in sideroblastic anemia and are often associated with Pearson syndrome, which includes pancreatic insufficiency and a degree of bone marrow failure.
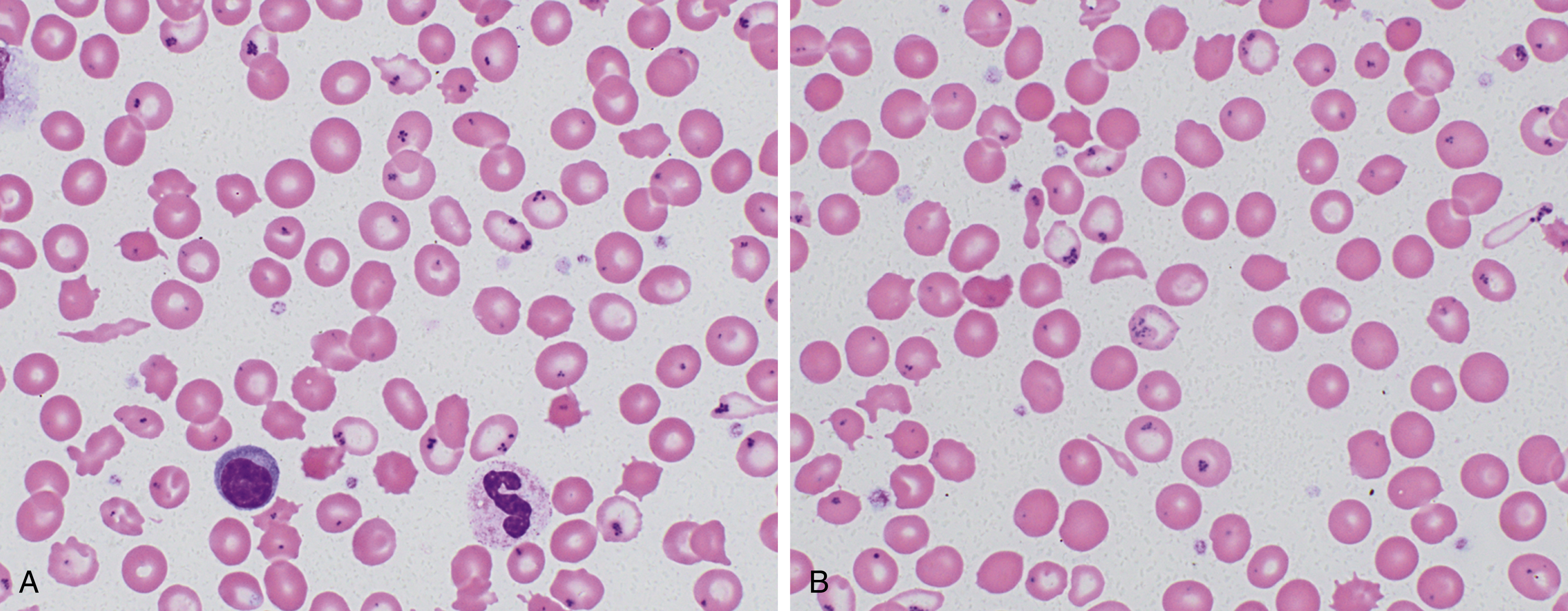
Hemolytic Anemias
Hemolytic anemia is caused by increased red blood cell destruction. The normal red blood cell life span is approximately 120 days, with senescent cells removed from the circulation by the reticuloendothelial system, predominantly in the spleen. This may happen in part through decreased sialic acid expression on the red blood cell membrane exposing an asialoglycophorin molecule that can be recognized by an autoantibody. , Accelerated red blood cell destruction may be caused by a range of congenital and acquired abnormalities. These include intrinsic defects in the red blood cell itself caused by abnormal membrane properties, defects in metabolism making them more susceptible to oxidative stress, or abnormal hemoglobin production. Alternatively, extrinsic factors may result in increased extravascular or intravascular hemolysis of otherwise normal red blood cells. No single laboratory test or clinical finding is sufficiently specific or sensitive to identify all cases of accelerated hemolysis. Nonetheless, all can be characterized by a shortened red blood cell life span. Direct measurement of red blood cell life span may be performed through a number of methods ( Table 3.4 ), albeit these may be used less frequently than other potential laboratory measures of hemolysis. Laboratory data, combined with morphologic evaluation of the peripheral smear and red blood cell indices, can be used to direct subsequent testing as necessary.
| Method | Process Measured | Method/Details | Specificity |
|---|---|---|---|
| Differential agglutination , | Survival of nonautologous cells: measure transfused O red blood cells over time | Type O cells administered to type A or B, agglutinate with anti-A or anti-B, transfused O will not agglutinate | Historical interest, cannot be used to measure autologous red blood cell survival |
| Population label: Radioactive chromium | Survival of mixed-age red blood cell population | Label patient red blood cells with 51 Cr, inject, serially monitor disappearance; complicated by loss of bound 51 Cr with time | Intrinsic or extrinsic factors, can test autologous cells |
| Population label: Biotinylation | Red blood cell life span | Label ex vivo with NHS-biotin, measure periodically by flow cytometry using fluorescent-conjugated avidin or streptavidin | Sensitive for whole red blood cell life span, covalently bound so label not lost with time |
| Cohort label (e.g., 15 N-glycine or other nonradioactive isotopes) | Survival of a cohort of red blood cells all the same age | Oral administration of 15 N-glycine, incorporated in hemoglobin | Label not confined to hemoglobin |
| Free hemoglobin | Red blood cell lysis | Released from red blood cells | Intravascular hemolysis |
| Lactate dehydrogenase | Red blood cell or tissue lysis | Released from red blood cells, cleared more slowly than hemoglobin | Intravascular hemolysis, ineffective hematopoiesis, released by other injured cell types |
| Carbon monoxide production | Heme catabolism | Heme catabolism the only source of endogenous CO | Short-term fluctuations in hemolysis; 20% of heme from non-heme sources |
Hypochromic and Hemolytic Anemias
The identification of hypochromic anemia with evidence of hemolysis generally narrows diagnostic consideration to a distinct subset of potential entities. The majority of hemolytic anemias in this setting are caused by thalassemias, with defects in hemoglobin synthesis resulting in an imbalanced production of hemoglobin chains. Hemoglobin is composed of a tetramer consisting of two α and two β chains, with the main form of adult hemoglobin (α 2 β 2 ) being designated as HbA ( Table 3.5 ). Embryonic and fetal hemoglobins have a higher affinity for oxygen, but are produced from homologous genes on the same cluster as their adult counterparts. These hemoglobins include the embryonic α-globin chain ζ and both embryonic (ε) and fetal (γA and γG) β-globin chains. Significant fetal hemoglobin (α 2 γ 2 , HbF) is present at birth. A minor adult β globin (δ) is also produced, designated HbA 2 .
| Hemoglobinopathy | HbA 2 | HbA | HbF | Hb Bart’s | HbH |
|---|---|---|---|---|---|
| α 2 δ 2 | α 2 β 2 | α 2 γ 2 | γ 4 | β 4 | |
| β-thalassemia | |||||
| Minor | 3.5% to 7% a | Present | 1% to 3% | — | — |
| Major | Variable | None if β 0 | 10% to 90% | — | — |
| α-Thalassemia | |||||
| α + | Not increased | Present | — | 1% to 2% (birth) | — |
| α 0 trait | Not increased | Present | — | 5% to 15% (birth) | — |
| HbH disease | Slightly decreased | Present | — | Trace | 5% to 40% |
| Hydrops fetalis | None if α 0 | None if α 0 | None if α 0 | Predominant form b |
a Concurrent iron deficiency anemia may preclude detection of elevated A 2 , creating confusion with α-thalassemia.
b 10% to 20% hemoglobin Portland (ζ 2 γ 2 ) may also be present with the embryonic α chain.
Defects causing one of the globin chains to be present in excess leads to protein precipitates within the red blood cell, resulting in ineffective erythropoiesis and reduced red blood cell life span from increased elimination in the spleen. α- and β-thalassemias are the two major categories of clinical significance, but abnormalities of other globin genes have also been described. While a cause of hemolysis and anemia, the mutations in heterozygous form offer a protective advantage against malaria. As a result, the prevalence of these mutations in certain endemic regions makes it one of the most common of all human genetic disease traits. ,
α-Thalassemia
α-Thalassemias show a worldwide distribution most prominent in Africa, the Mediterranean, the Middle East, and Southeast Asia, but the populations are affected by different mutational patterns ( Table 3.6 ). Multiple mutations appear to have arisen and conferred a survival advantage. The α-globin cluster (chromosome 16) includes one ζ gene and two α genes (designated α 1 and α 2 ) along with four pseudogenes. Inherited mutations in α-thalassemia may affect one or both α genes with the resultant inheritance of zero to four functional α genes from both parents. Variants with loss of both α-globin chains on a single chromosome (α 0 ) are most common in Mediterranean and Asian populations. When individuals are homozygous for α 0 , they have a complete lack of α chain production and hemoglobin Bart’s hydrops fetalis, where hemoglobin Bart’s is the γ tetramer (γ 4 ) (see Table 3.5 ). Even without complete deficiency, hydrops fetalis may occur when α-globin gene production drops to less than approximately 25% of normal values.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


