Hormones and Cancer
20.1 INTRODUCTION
Breast and prostate cancers are the most commonly occurring cancers in Western society, and are the second leading cause of cancer death (next to lung cancer) in women and men, respectively. Both of these cancers arise in tissues that require steroid sex hormones (estrogens and androgens) for their development, growth, and function. Although human cancers occur in other hormone-dependent tissues, such as the uterus, ovary, and testis, this chapter focuses exclusively on breast and prostate cancer as models of hormone-dependent cancers.
The relationship between prostate enlargement and hormones produced by the testes has long been recognized. Although the chemical nature of androgens was not known, it was reported in 1895 that castration of elderly men with prostate enlargement, presumably as a result of benign prostatic hyperplasia, resulted in rapid atrophy of prostatic tissue. Early anecdotal evidence regarding the testicular (ie, androgen) dependence of the human prostate is also derived from studies involving eunuchs from the Ottoman and Chinese courts, which indicated that prostates did not develop in prepubertal castrates. Following the isolation of “testosterone” as the most potent androgenic compound in the testes in 1935, Huggins and Hodges demonstrated the efficacy of surgical orchiectomy for the treatment of metastatic prostate cancer, for which Huggins received the Nobel Prize for Medicine in 1966. Similarly, a link between estrogen and breast cancer growth was established at the end of the 19th century, when Beatson demonstrated that oophorectomy was useful in the treatment of metastatic breast cancer in some premenopausal women. However, a molecular basis for this observation was not forthcoming until the 1960s with the discovery of the estrogen receptor, followed by the demonstrated expression of estrogen receptors in some human breast tumors by Elwood Jensen.
Evidence for a direct link between the action of sex steroids, and a causal role in the carcinogenic process leading to breast and prostate tumors, was first provided by Robert Noble. He reported that prolonged exposure to estrogen, androgen, or combinations of the 2 led to breast and prostate cancers in rats. More recently, the successful use of the antiestrogen, tamoxifen, to reduce the incidence of breast cancer in high-risk women, supports a direct link between estrogen action and breast tumor formation. These hormones are fundamental not only to the development of normal mammary and prostate glands, but also to dysplastic and neoplastic processes that occur in these tissues.
In this chapter, relationships between hormones and cancers of the breast and prostate are explored in the context of basic mechanisms of hormone action, the natural history of the 2 diseases, and their treatment with hormonally based therapies.
20.2 BASIC MECHANISMS OF HORMONE ACTION
Hormones can be classified generally into 2 broad groups: (a) nonsteroidal (amino acids, peptides, and polypeptides), which usually require cell-membrane localized receptors that regulate second messenger molecules such as cyclic adenosine monophosphate (cAMP) to mediate their action (see Chap. 8, Sec. 8.2), and (b) steroidal, which bind directly to intracellular receptors to mediate their action. Breast and prostate cancer are dependent primarily on estrogen and androgen steroid hormones, respectively, for their growth and viability. Other examples of steroid hormones include glucocorticoids, miner-alocorticoids, and progestins such as progesterone.
The bioavailability of steroid hormones at the site of action depends on several factors, including synthesis, transport via the blood, access to target tissue, metabolism in target tissue, and expression of receptors within the target cell.
20.2.1 Synthesis and Metabolism of Estrogens
As indicated in Figure 20–1, all steroids are synthesized from the common precursor, cholesterol. The primary site of synthesis of estrogens (eg, estrone and estradiol) in premenopausal women is the parafollicular region of the ovary. Ovarian steroid synthesis in premenopausal women is cyclical and regulated via the gonad–hypothalamus–pituitary feedback axis, as indicated in Figure 20–2A. Other sites of estrogen biosynthesis include mesenchymal cells in adipose tissue and skin; these tissues become major sources for estrogen synthesis in postmenopausal women where adrenal androgens, in particular androstenedione, are converted to estrone by aromatase cytochrome P450 (Simpson, 2000). Some estrone molecules can then be converted to the more potent estradiol-17β by 17β-hydroxysteroid dehydrogenases (Miettinen et al, 2000). A large amount of estrone is converted by estrone sulfotransferase to estrone sulphate, which has a longer half-life in blood, and therefore can act as an estrogen reservoir, being converted back to estrone by the action of sulfatases (Miettinen et al, 2000). Aromatase activity has been detected in normal human breast tissue and in more than 50% of human breast tumors (Sasano and Harada, 1998). Estrone sulfotransferase, sulfatase and 17β-hydroxysteroid dehydrogenase type 1 have been detected in both normal and malignant human breast tissues (Miettinen et al, 2000) and the relative expression of these enzymes probably regulates the local availability of estrogens to target cells. Estrogen synthesis in postmenopausal women is not cyclical but serum and local tissue levels can differ between individuals because of a variety of environmental and genetic factors such as obesity and genetic variation/polymorphism in steroid metabolizing (biosynthetic and degrading) enzymes (Thompson and Ambrosone, 2000).
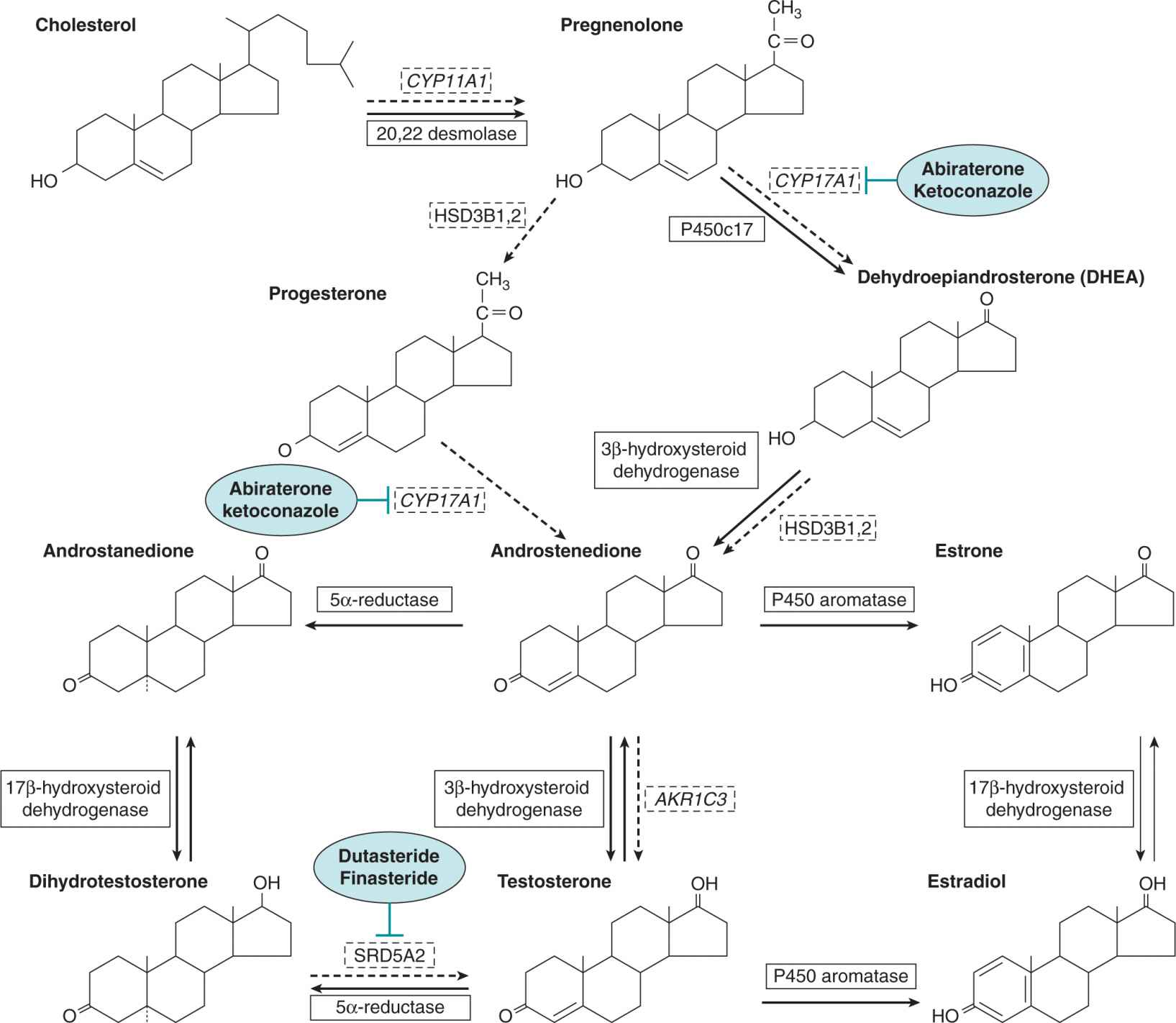
FIGURE 20–1 The principal mammalian steroid biosynthetic pathways for estradiol, testosterone, and dihydrotestosterone are represented by solid arrows. The enzymes that catalyze the reactions are shown in boxes. Although the main sites for steroid synthesis are the gonads and the adrenals, metabolic interconversions and activations occur in sex hormone target tissues, such as the prostate and breast, as well as other peripheral tissues, such as skin and adipose tissue. Dashed arrows and boxes represent the intratumoral androgen synthesis pathway. Inhibitors of enzymes are represented in blue ovals.
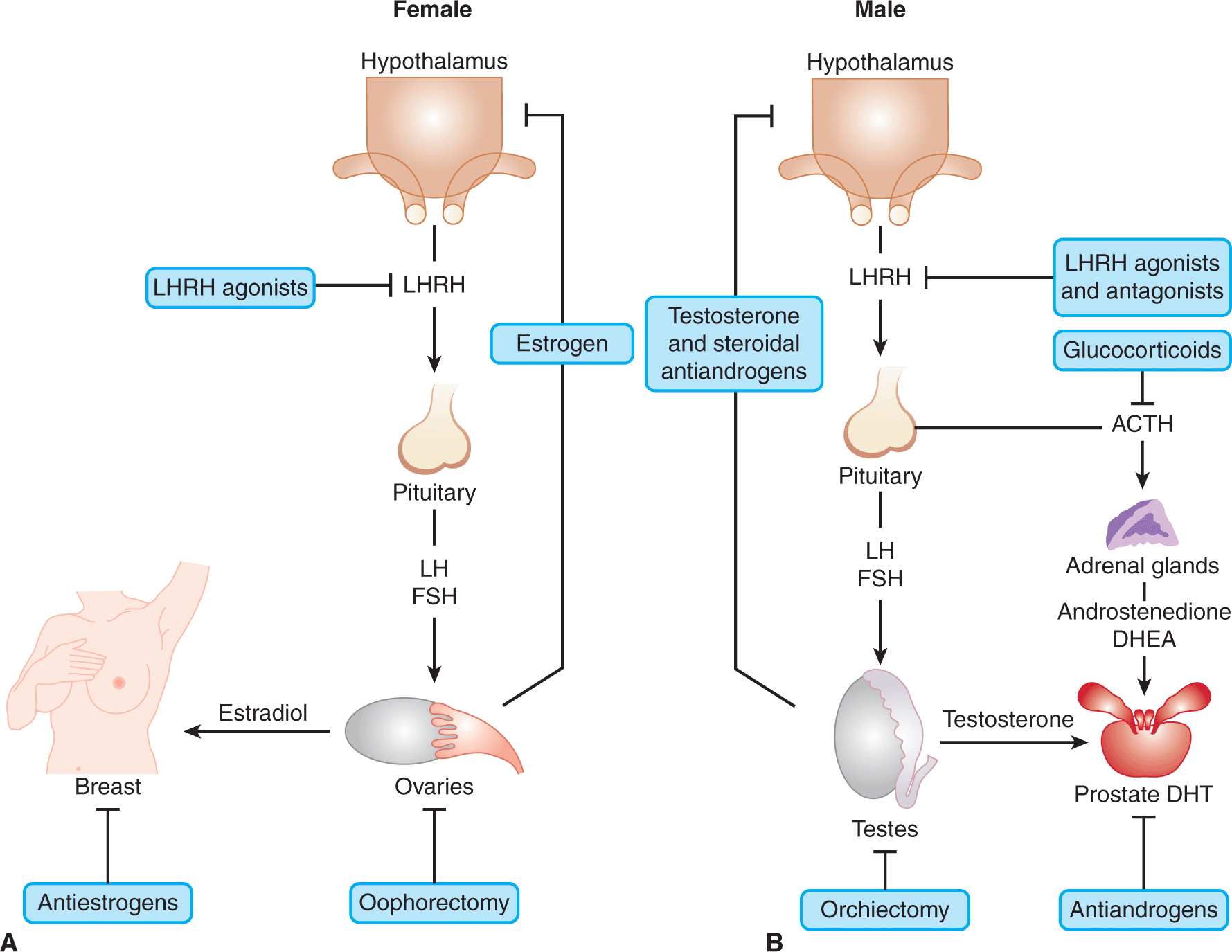
FIGURE 20–2 The gonadal–hypothalamic–pituitary axis. The pathways and feedback loops that regulate the production of estrogens in females (A) and androgens in males (B) and their target tissues are shown. The procedures and agents used for blocking the synthesis and activity of androgens and estrogens at the various steps in the pathway are highlighted. Note that luteinizing hormone-releasing hormone (LHRH) agonists initially stimulate release of luteinizing hormone (LH) followed by its inhibition. ACTH, adrenocorticotropic hormone; FSH, follicle-stimulating hormone; DHEA, dehydroepiandrosterone; DHT, dihydrotestosterone.
20.2.2 Synthesis and Metabolism of Androgens
The Leydig cells of the testes produce almost 90% of the body’s androgens with the remainder being made mainly by the adrenal cortex. As illustrated in Figure 20–2B, the testes make primarily testosterone, whereas the major adrenal androgens are dehydroepiandrosterone (DHEA) and its derived sulfate, which although weak androgens, can be converted in other tissues to testosterone. The principal circulating androgen in man is testosterone (see Fig. 20–1). As with estrogen synthesis in the ovary, testosterone production in the testis is regulated by a negative feedback loop involving luteinizing hormone (LH) and luteinizing hormone-releasing hormone (LHRH) via the gonad–hypothalamus–pituitary feedback axis (see Fig. 20–2B). Although there are diurnal fluctuations of androgen secretion, their production does not follow a regular cyclical pattern. Also, there is no apparent equivalent to the menopause in men, although there is a progressive decrease in testosterone levels with age, which is accompanied by some degree of testicular failure.
There are 2 principal pathways through which testosterone can undergo metabolic conversion to more potent forms (see Fig. 20–1). The first involves the enzyme aromatase, which is present in many tissues (eg, adipose tissue, testis, brain) and which can convert approximately 0.5% of the daily production of testosterone to estradiol. Although this is a small proportion of the total amount, estradiol is a 200-fold more potent inhibitor of gonadotrophins than testosterone. The second major pathway is the conversion of testosterone to the more potent androgen dihydrotestosterone (DHT) by the enzyme 5α-reductase, which is present in many androgen target tissues such as the prostate and skin. There are 2 isoforms of 5α-reductase, each with different kinetic properties and sensitivity to inhibitors, with the type II isoform predominating in human accessory sex tissue (Bruchovsky et al, 1988). The interaction of testosterone and DHT with the androgen receptor is different. Testosterone has a 2-fold lower affinity than DHT for the androgen receptor and is more effective in regulating differentiation. However, the dissociation rate of DHT from the receptor is 5-fold slower than testosterone, and it is a more potent androgen to induce growth and maintenance of the prostate gland (Grino et al, 1990).
20.2.3 Transport of Steroid Hormones in the Blood
As hydrophobic molecules in an aqueous environment, most steroid hormones are transported in the blood bound to proteins, predominantly sex hormone-binding globulin (SHBG) and albumin (as shown in Fig. 20–3). Only approximately 2% is in an unbound form, which is the biologically active fraction. SHBG also plays a role in permitting certain steroid hormones to act without entering the cell: estrogens and androgens bind with high affinity to SHBG, which, in turn, interacts with a specific, high-affinity receptor (SHBG-R) on cell membranes and transduces a signal via a G-protein/cAMP (Kahn et al, 2002). The steroid/SHBG-R/SHBG complex generates messages that have effects on the transcriptional activity of regular, intracellular receptors for steroid hormones. Hence, SHBG not only modulates the amount of ligand available to bind to these steroid receptors, but may also modulate their activity through interaction with the cell membrane. Factors that influence the levels of SHBG and albumin will affect the bioactivity of steroid hormones. For example, SHBG production is stimulated by both estrogens and by the antiestrogen tamoxifen, whereas androgens and progestins have been shown to suppress it. In addition, SHBG can be expressed inside the cytoplasm of prostate cells from de novo synthesis and, through binding to steroids such as DHT, can affect the intracellular concentration of free androgens (Kahn et al, 2002).
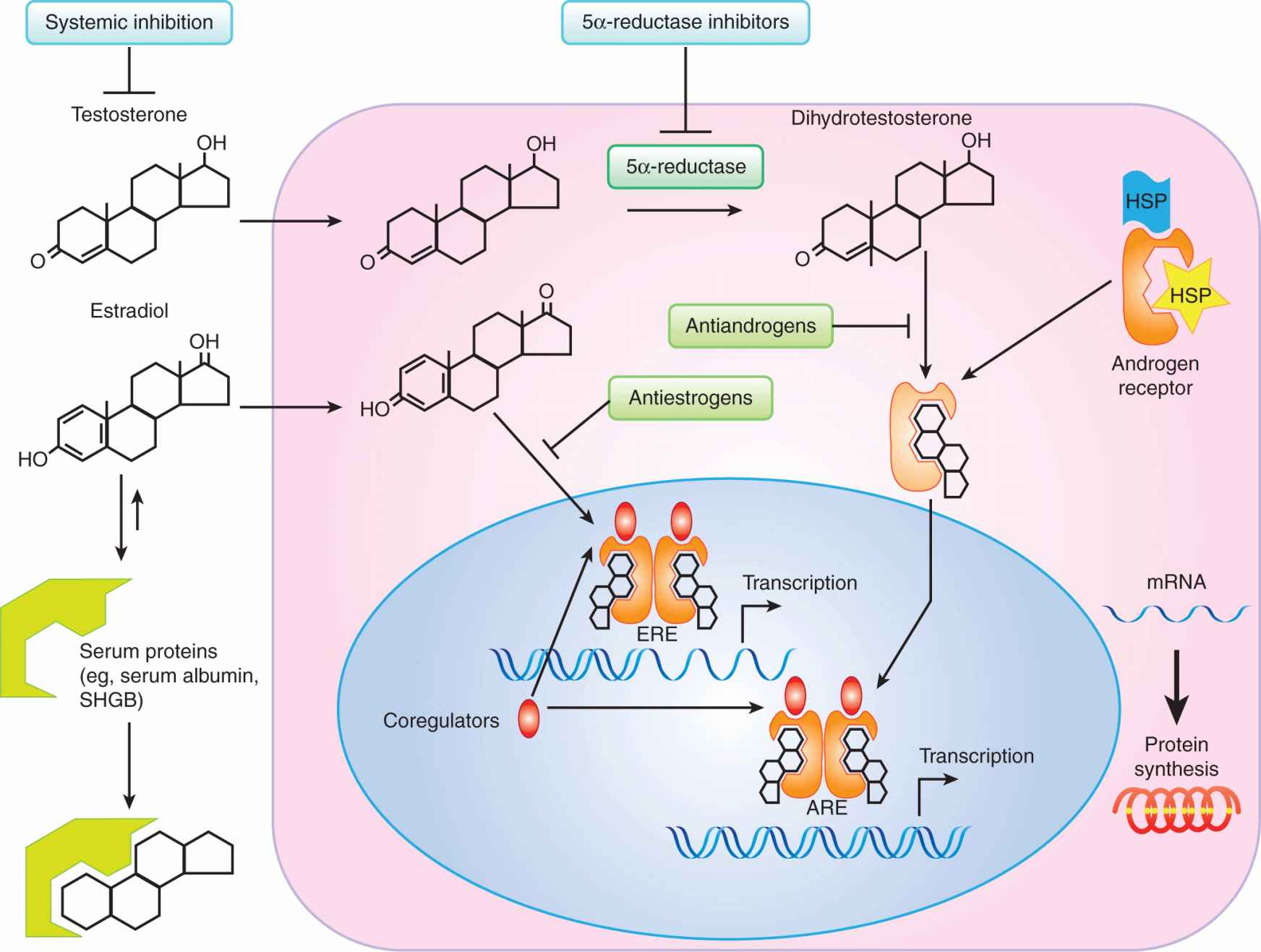
FIGURE 20–3 Schematic pathways for the molecular action of testosterone and estradiol in hormone target cells. A relatively simplistic overview of the key events in steroid hormone action with the sites at which the hormone signal can be inhibited, are indicated. Although the dynamics of estrogen and androgen action are similar, for full activity testosterone needs to be converted to dihydrotestosterone, which binds to a cytoplasmic form of the androgen receptor and is translocated into the nucleus, whereas estradiol binds directly to its receptor in the cell nucleus. ARE, androgen-responsive element; ERE, estrogen-responsive element; HSP, heat shock protein; SHBG, sex hormone-binding globulin.
20.2.4 Steroid Hormone Receptors
An overview of how steroid hormones regulate growth and differentiation of their target cells is shown in Figure 20–3. The majority of steroid hormone that enters the cell is derived from the small nonbound fraction in the circulation, which can enter by passive diffusion. Upon entry into the cell, the steroid or its metabolic derivative (eg, DHT) binds directly to a predominantly cytoplasmic (androgen receptor) or nuclear (estrogen receptor) steroid receptor protein. The steroid-receptor complex undergoes an activation step involving a conformational change and shedding of heat shock (including HSP70, HSP90, and HSP40) and other chaperone proteins, which are necessary to maintain the receptor in a competent ligand-binding state (Aranda and Pascual, 2001). After dimerization, and nuclear transport in the case of some steroid receptors like the androgen receptor, the activated receptor dimer complex binds to specific DNA motifs called hormone-responsive elements (HREs) found in the promoters of hormone-regulated genes (Aranda and Pascual, 2001). The receptor-DNA complexes, in turn, associate dynamically with coactivators and basal transcriptional components to enhance the transcription of genes, whose messenger RNAs (mRNAs) are translated into proteins that elicit specific biological responses. It is likely that receptors that are not bound to their ligands, or those bound to antagonists (Fig. 20–4), form complexes with core-pressors to inhibit the transcription of specific genes (Perissi and Rosenfeld, 2005).

FIGURE 20–4 Illustration of the role of coactivators and corepressor in regulation of steroid receptor action. In the presence of agonistic ligands (eg, estradiol for estrogen receptor [ER] and DHT for androgen receptor [AR]), the steroid receptor-DNA complexes associate dynamically with coactivators, which, in turn, recruit other proteins, including cointegrator complexes, that contact and stabilize the basal transcription unit resulting in enhanced transcription of target genes. In the presence of antagonistic ligands (eg, tamoxifen for ER and flutamide for AR), the receptors are in a different conformational state (steroid receptor ligand analog) and the receptor-DNA complexes dynamically associate with corepressors (Co-R) which destabilize basal transcription units and result in reduced transcription of target genes. HRE, hormone-responsive element.
All steroid receptors are members of a so-called superfamily of more than 150 proteins. All are ligand-responsive transcription factors that share similarities with respect to their structural homology and functional properties. As shown in Figure 20–5, each member of the steroid receptor family possesses a modular structure composed of the following:
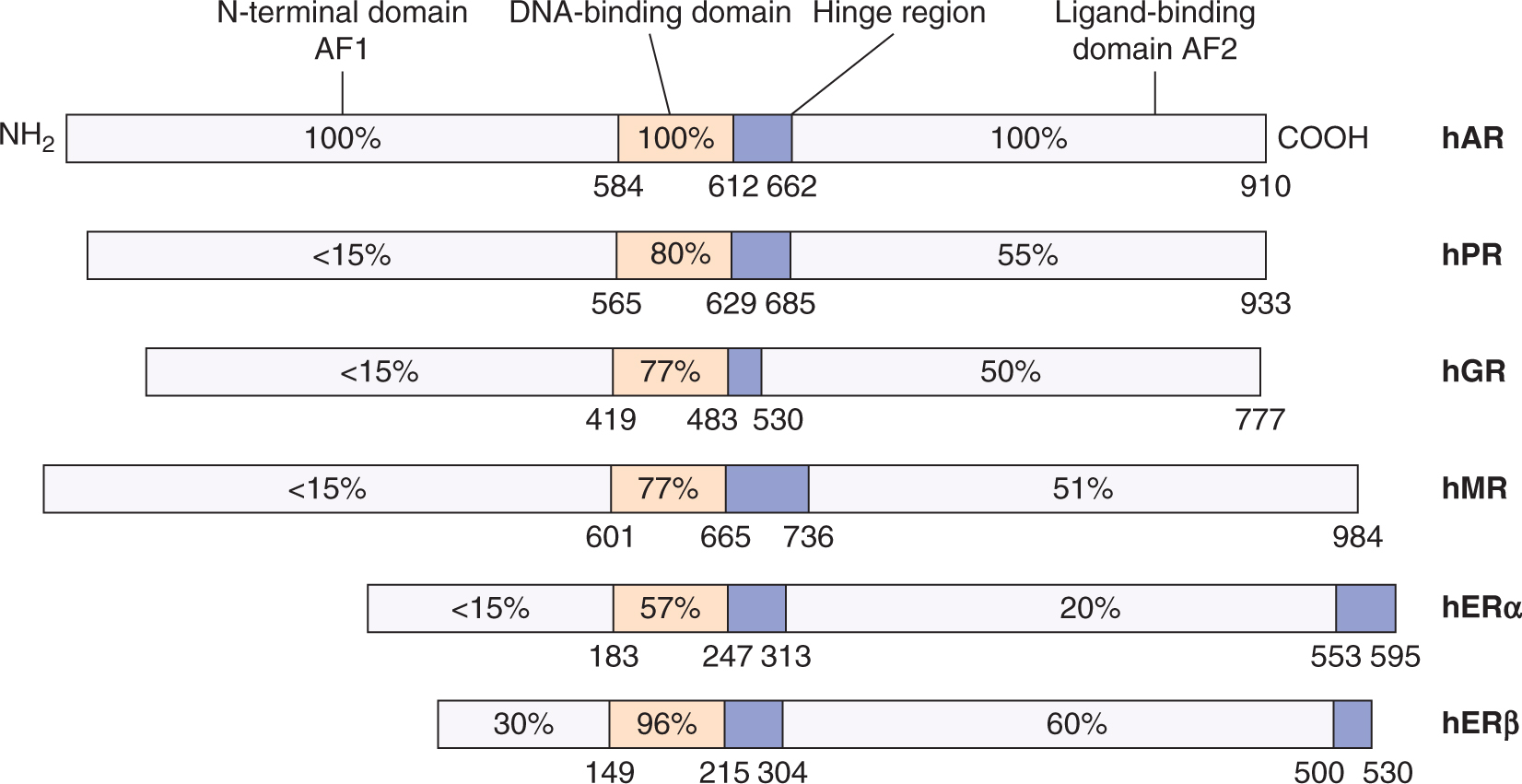
FIGURE 20–5 The relative amino acid sequence homology within the functional domains of the principal members of the family of human (h) steroid hormone receptors. The relative amino acid sequence homology (represented by the percentages indicated in boxes) for hERβ is in reference to hERα, whereas all the other receptors are relative to hAR. The numbers correspond to the amino acid positions from the N-terminus (NH2), AF1 and AF2 refer to the transcriptional activation function 1 and 2 that reside in the N-terminal and ligand-binding domains, respectively. AR, Androgen receptor; ER, estrogen receptor; GR, glucocorticoid receptor; MR, mineralocorticoid receptor; PR, progestin receptor.
1. An N-terminal region containing ligand-independent transcriptional activating functions (collectively referred to as activating function-1 or AF1);
2. A centrally located DNA binding domain of approximately 65 amino acids having 2 zinc fingers (see Chap. 8, Sec. 8.2.6);
3. A hinge region that contains signal elements for nuclear localization; and
4. A ligand-binding domain in the C-terminal region of the protein containing a ligand-dependent transcriptional activating function (called AF2).
AF1 and AF2 can function independently or synergistically, depending on the gene promoter and/or the cell type (Aranda and Pascual, 2001). Between members of the family of steroid receptors, the N-terminal region has the highest degree of amino acid sequence variability, whereas the DNA-binding domain has the most shared homology.
20.2.4.1 Androgen Receptors The androgen receptor (AR) is encoded on the X chromosome. As a single allele gene in males, it is susceptible to genetic defects whose phenotypes can range from minor undervirilization to a complete female phenotype known as testicular feminization. The normal, wild-type AR has a molecular weight of approximately 110 kDa, but truncated molecular variants have been observed in a variety of prostate cancers. These variants lack the ligand-binding domain of the AR but are still functional, even in the absence of androgen (Hu et al, 2009). In this chapter, AR refers only to the wild-type, 110-kDa form.
Relative to other steroid receptors, the AR has one of the largest N-terminal domains, which occupies more than half of the primary sequence of 919 amino acids (see Fig. 20–5). A unique feature of the AR relative to other members of this family is the occurrence of several stretches of the same amino acids (termed homopolymeric) in the N-terminal domain. These tracts include 17 to 29 repeating glutamine residues, starting approximately at amino acid 59, 9 proline residues at amino acid 372, and a 24-residue polyglycine stretch beginning at amino acid 449. These repeating tracts have been shown to modulate the folding and structural integrity of the N-terminal domain (Davies et al, 2008) and there is evidence of a reverse relationship between variations in their lengths and the transcription levels of mRNAs (Robins et al, 2008). Not surprisingly, these variations result in biological effects: for example, an abnormal extension of the polyglutamine tract to 40 or more residues is associated with neurodegenerative diseases such as X-linked spinal and bulbar muscular atrophy (La Spada et al, 1991). In contrast, a decreased size of these homopolymers may be linked with the development of prostate cancer (Robins et al, 2008), although there is no consensus on this relationship (Mir et al, 2002).
20.2.4.2 Estrogen Receptors There are 2 estrogen receptor (ERs), ERα and ERβ, which, unlike AR isoforms, are encoded by separate genes (see Fig. 20–5). Several variant isoforms of each ER, generated by alternative RNA splicing, may also be expressed (Murphy et al, 1998). The centrally located DNA-binding domains for ERα and ERβ (see Fig. 20–5) are highly homologous (>95% identity). Their C-terminally located ligand-binding domains are approximately 60% identical. As with other steroid receptors, the ligand-binding domain contains a ligand-dependent dimerization function, and a ligand-dependent transactivation function, AF2. Similarly, a ligand-independent transactivation function, AF1, is present in the N-terminal domain. This latter domain is different in ERα and ERβ. In addition steroid receptors are subject to multiple posttranslational modifications such as phosphorylation, which can regulate receptor activity, transcriptional activity, DNA binding, protein turnover, and ligand sensitivity (Ward and Weigel, 2009).
20.2.5 Binding of Steroid Receptors to DNA
As transcription factors, steroid receptors modulate gene expression by first binding to specific DNA sequences in the promoter regions of hormone-regulated genes (see Fig. 20–4). The α-helix structure of the first zinc finger (see Chap. 8, Sec. 8.2.6) of the DNA-binding domain of a receptor is the primary discriminator for binding to different DNA sequence motifs of HREs (Aranda and Pascual, 2001). All members of the nuclear receptor family bind as dimers to pairs of a similar DNA sequence motif, AGNNCA (N = any nucleotide), which comprises the HRE found in the promoters of steroid regulated genes (Glass, 1994). Steroid receptors can bind to multiple HREs that are often hundreds of kilobase pairs distal to target gene promoters; furthermore steroid receptors and associated complexes bound at both distal and proximal HREs can interact, resulting in chromosome looping, to regulate target gene transcription (Fullwood et al, 2009). Nuclear receptors can be further subdivided on the basis of selection of the primary sequence of the core motif as either AGGTCA, for the ER and thyroid receptor subfamily, or AGAACA for the AR, glucocorticoid receptor (GR), or progestin receptor (PR). In general, 2 or more sets of interacting HREs in a gene promoter are required to elicit a steroid-mediated response (Klinge, 1999; Rennie et al, 1993).
The very high sequence homology of HREs has raised the question as to how steroid receptors govern hormone-specific responses. Although there is no definitive mechanism, the interaction with unique combinations of coregulators or other binding proteins is probably a major contributing factor to steroid-receptor specific gene regulation. Other parameters may also be important, such as the availability of receptor and ligand, the activity of proximal transcription factors, the cooperative binding of receptors to 2 or more DNA-binding sites, and altered DNA target motif recognition.
20.2.6 Interaction of Steroid Receptors with Coregulator Proteins
Important molecular mechanisms by which nuclear receptors regulate gene transcription involve not only direct interactions of steroid receptors with some of the basal transcription components, but also indirect interactions through recruitment of coregulator protein complexes to the promoters of target genes (Perissi and Rosenfeld, 2005; see Figs. 20–3 and 20–4). Coregulators fall into 2 main classes, coactivators and corepressors, which enhance or repress transcription, respectively (as illustrated in Fig. 20–4). There are many genes that encode coactivators, but the steroid receptor coactivator (SRC)/NCOA/p160 family are relatively specific for nuclear receptors and are the best studied (O’Malley and Kumar, 2009; Xu et al, 2009).
Although the auxiliary molecular components involved and the dynamics of their interactions remain to be established, a general pattern has emerged. The coactivators and corepressors are important for mediating both AF2 and AF1 activities of steroid receptors. Their mechanism of transcriptional activation is thought to involve 2 stages (Perissi and Rosenfeld, 2005). First, when recruited to the receptor by direct protein–protein binding, coregulators promote the local remodeling of chromatin structure through acetyltransferase or deacetylase activity and through their ability to recruit other proteins with chromatin remodeling activity (as outlined in Fig. 20–6). Second, the coactivators recruit and/or stabilize the basal transcription machinery by protein–protein interactions so as to enable efficient transcription of the target gene by RNA polymerase II (Perissi and Rosenfeld, 2005). The chromatin remodeling enables altered access of the promoter DNA to general transcription factors (Fig. 20–6).
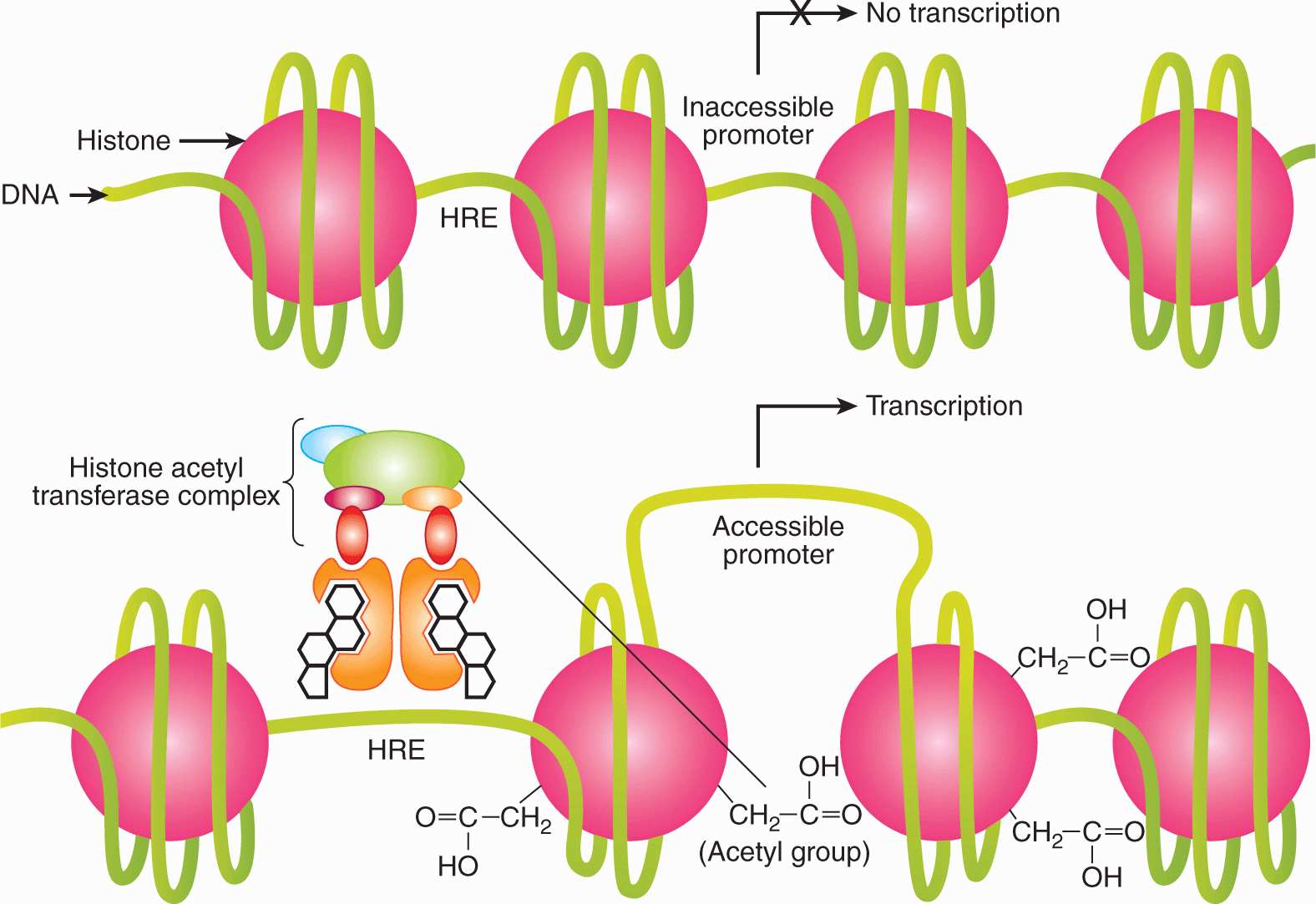
FIGURE 20–6 Remodeling of chromatin and activation of transcription by steroid hormone receptors. Steroid hormone receptors bind to hormone responsive elements (HREs) in the promoters of target genes and recruit coactivators (eg, SRC-1) and cointegrators (eg, CBP/p300), which have chromatin remodeling activity. Some coregulators and cointegrators have histone acetyltransferase (HAT) activity, which results in dynamic nucleosomal histone acetylation and increased access of basal transcription complexes (that include RNA polymerase II) to the promoters of target genes.
It is uncertain as to which coactivators are necessary or sufficient for transcriptional activation. In MCF-7 breast cancer cells ERα and a number of coactivators associate rapidly with target promoters in a dynamic, cyclic fashion and the SRC/NCOA/p160 class of coactivators is sufficient for gene activation (Shang et al, 2000). It is likely that the relative availability of coregulators will vary in different tissues and may even be restricted to specific tissues. Also, although the occurrence of receptor-specific coregulators has not been confirmed, many coregulators bind preferentially to certain receptors (Leo and Chen, 2000). For example, a repressor of ER transcriptional activity, called repressor of estrogen receptor activity (REA) is active on ERα or ERβ, but not other steroid receptors (Montano et al, 1999). Its mechanism of action involves a competition with coactivators such as SRC-1 for binding to the ligand-binding domain of ER (Delage-Mourroux et al, 2000). Furthermore, a member of the SRC/NCOA family, SRC-3/NCOA3 or amplified in breast cancer 1 (AIB1) may have a role in breast cancer as it is amplified and overex-pressed in some breast tumors and such overexpression has been correlated with tamoxifen resistance (Anzick et al, 1997; Xu et al, 2009).
20.2.7 Mechanisms for Transcriptional Regulation by Steroid Receptors
There are at least 3 different mechanisms by which steroid receptors regulate transcription of target genes:
1. Ligand-dependent and requiring direct binding of steroid receptors to HREs in promoter DNA (as described above and illustrated in Figs. 20–3 and 20–4);
2. Ligand-dependent but not requiring direct binding to DNA; and
3. Ligand-independent action.
Most hormone-regulated genes have ligand-dependent binding to HREs. Examples of target genes regulated in this fashion are prostate-specific antigen (PSA) and prostate-specific membrane antigen (PSMA). However, in some target genes, ligand-activated ER regulates transcription, without contacting the DNA directly, through protein–protein interaction with other transcription factors that are in direct contact with DNA via their own specific response elements. For example, ERα can interact with Sp1 and AP-1 transcription factors and regulate transcription of some genes.
Ligand-independent activation of ER and AR can occur through crosstalk with a variety of growth factor networks. Growth factors such as epidermal growth factor (EGF) and/or insulin-like growth factor (IGF)-I bind to their respective tyro-sine kinase receptors located in the plasma membrane of target cells and activate signal transduction pathways involving activation of other kinases (see Chap. 8, Sec. 8.2); these events can lead to phosphorylation of steroid receptors. One enzyme activated by growth factor signaling that can phosphorylate directly both ERs as well as the AR is mitogen activated protein kinase (MAPK) (Ueda et al, 2002). Similarly, interleukin-6 and protein kinase A can activate AR and ERα directly in the absence of the appropriate steroid ligand. Interleukin-6 may bind to and influence AR activity without inducing phosphorylation of the AR (Ueda et al, 2002). Growth factor/phosphorylation pathways can also influence steroid hormone receptor pathways via their ability to modulate coactivators by phosphorylation (Han et al, 2009). These alternative pathways for regulation of ER and AR activity could have a profound influence on the emergence of hormone independence in tumors that have not lost their hormone receptors (see Sec. 20.5.5).
20.2.8 Nontranscriptional Actions of Steroid Receptors
There is evidence that not all effects of steroid hormones are mediated via the regulation of genomic or transcriptional events (Kelly and Levin, 2001). Transcription-independent effects of estrogen manifest themselves as rapid responses in target cells of the order of seconds to a few minutes. For estrogen, examples of nongenomic effects include modulation of calcium ion flux, effects on membrane channels in the central nervous system and peripheral excitable cells, membrane-associated interactions with growth factor receptors, and interactions with survival/apoptosis pathways. There is evidence that membrane-associated ERs coupled to G proteins or nitric oxide-generating systems mediate some of these actions. At least 2 categories of membrane-based receptors may exist: one related to the classical intracellular ERα or ERβ (Kelly and Levin, 2001), and the other distinct from them (Nadal et al, 2000). Other steroid hormones such as progesterone and testosterone may also influence cells via related mechanisms (Kousteni et al, 2001).
The importance of nontranscriptional actions of estrogen in breast cancer in relation to estrogen-dependent signaling is not known. There are multiple levels of estrogen interaction with growth factor receptor kinases and the signal transduction pathways that they regulate. Therefore, nongenomic and genomic actions of estrogen are likely to be integrated with and complementary to each other. Unravelling this molecular complexity has important implications with respect to new therapeutic combinations and approaches (Arpino et al, 2008).
20.2.9 Quantification of Steroid Hormone Receptors
ER and PR are biomarkers in breast cancer. In particular, they help to predict the likelihood of response to endocrine therapy. In an unselected group of breast cancer patients with advanced disease, 30% to 40% will respond to endocrine therapy. However, patients whose tumors are both ER and PR negative have less than 10% chance of responding to endocrine therapy while in patients selected for the presence of both ER and PR in their primary breast tumor, the response rate to endocrine therapy is 70% to 80%.
Approximately 90% of unselected men with prostate cancer will respond to endocrine therapy: AR provides no prognostic or diagnostic value since almost all tumors usually possess a functioning AR, and almost all men respond initially to androgen withdrawal.
ER and PR are measured routinely in breast tumor biopsies by immunohistochemistry (IHC) using well characterized, specific monoclonal antibodies, as outlined in Figure 20–7A. IHC methods (Fitzgibbons et al, 2010; Hammond et al, 2010) can determine whether the detected receptor is within tumor cells, and can show receptor heterogeneity in tumor tissue. An example of ER heterogeneity within a breast tumor is shown in Figure 20–7B. Heterogeneity refers to the observation that both ER+ and ER– breast cancer cells can be present to varying degrees within any breast cancer biopsy sample, in addition to the presence of other types of cells (vascular cells, infiltrating cells of the immune system, normal stromal fibroblasts, normal breast adipocytes, and normal breast epithelial cells). Semiquantitative methods have been used to evaluate ER and PR by IHC, but positive results are generally reported when more than 1% of tumor nuclei stain positively (Hammond et al, 2010). There is a correlation between benefit and increasing ER level, which can be achieved by combining the level of staining intensity with the percentage of tumor cells positively stained to give H-scores or Allred scores (Hammond et al, 2010). Results can vary amongst laboratories, especially in the low to middle range of the receptor spectrum, and guidelines for standard operating procedures for tissue collection, assay validation, quality control, and interpretation have been published (Fitzgibbons et al, 2010; Hammond et al, 2010).
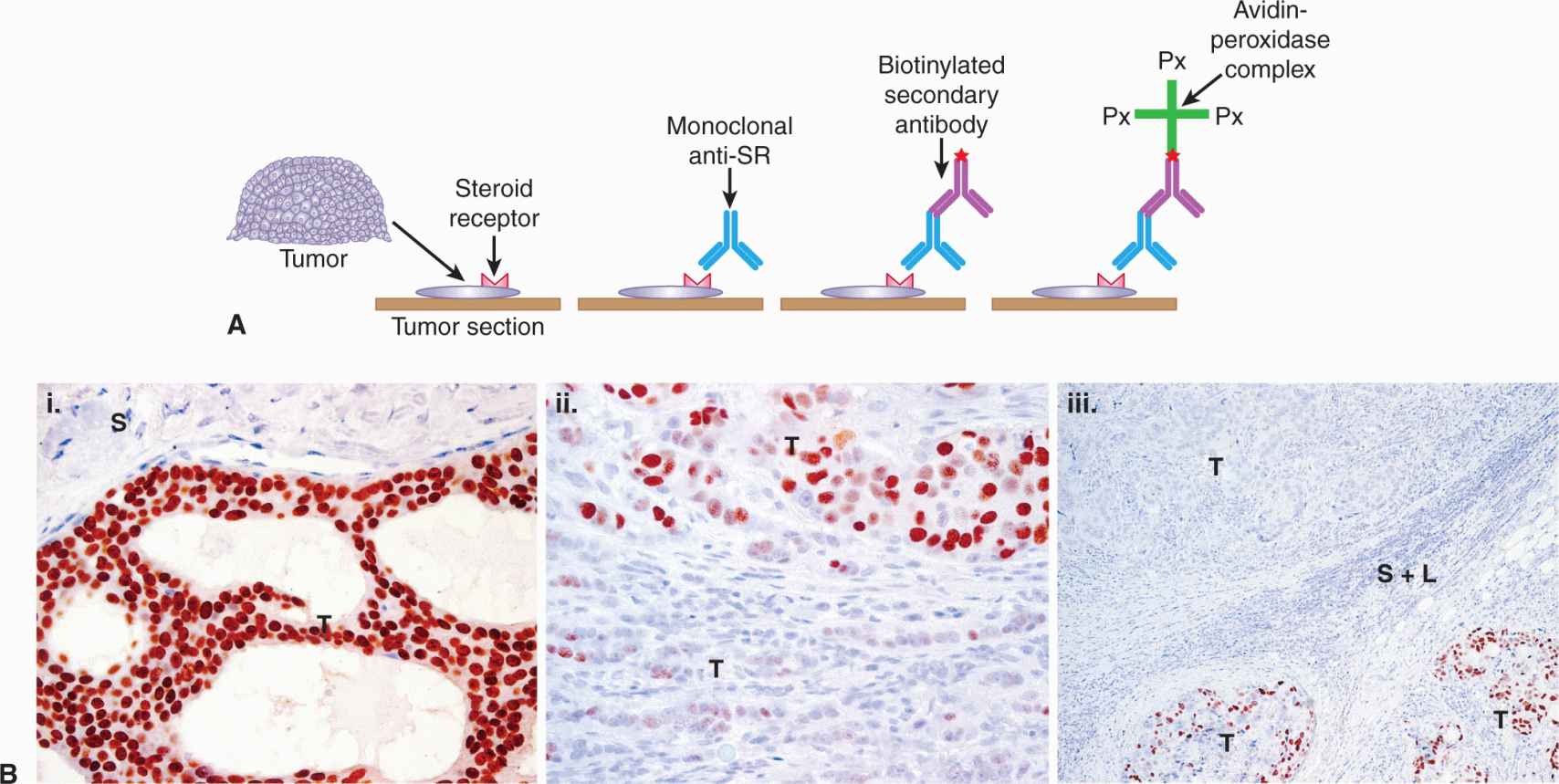
FIGURE 20–7 Immunohistochemical–Avidin-biotin complex method. A) Determination of hormone receptors in tumor tissue. Thin sections of tumor are cut from formalin-fixed, paraffin-embedded biopsy specimens. The section is next exposed to a monoclonal antibody specific for the steroid receptor (SR) being assessed. The section is then exposed to a second biotinylated antibody specific for the first antibody. Finally, avidin-peroxidase complex is added, followed by a chromogen, and color appears where the SR is located. B) Immunohistograms illustrating heterogeneity of human breast cancer biopsy samples. Brown staining represents ERα positivity. T, Invasive breast cancer; S, stromal and connective tissue elements; L, lymphocytes. (i) Homogenous expression of ERα within an invasive breast cancer, with negative adjacent vessels and stroma. (ii) Moderate heterogeneity of expression of ERα within an invasive breast cancer, with strong expression within solid nests of tumor cells in the upper field and weak or negative expression within less-cohesive clusters of tumor cells in the lower field. Stromal and lymphocytic elements are negative for ERα. (iii) Marked heterogeneity of expression of ERα within an invasive tumor metastatic to an axillary lymph node. In the upper part of the section one metastatic component is homogenously ERα –ve and in the lower part of the section the other component is moderate to highly ERα +ve. These 2 different elements are separated in this field of view by a band of fibrous stroma and infiltrating lymphocytes.
ER and PR can also be assayed by measuring their mRNA. This is currently being assessed as part of a 21-gene expression analysis, known as Oncotype DX, using reverse-transcription and quantitative polymerase chain reaction technology (see Chap. 2, Sec. 2.2.5). This evaluation is only carried out by the company that developed the assay, which generates a recurrence score (RS) that helps predict the benefit of adding chemotherapy to hormonal therapy in women with ER+ breast cancer (Paik et al, 2006; Albain et al, 2010). The clinical utility of the Oncotype DX assay and other multigene assays is still being evaluated in prospective clinical trials (TAILORx [Trial Assessing Individualized Options for Treatment for Breast Cancer]; MINDACT [Microarray in Node-Negative and 1-3 Node Positive Disease May Avoid Chemotherapy Trial]) (Cardoso et al, 2008).
20.3 NATURAL HISTORY OF BREAST AND PROSTATE CANCER
20.3.1 Risk Factors for Breast Cancer
Female gender and increasing age are the major risk factors for human breast cancer. Also, factors that increase the cumulative exposure and/or sensitivity of the breast epithelium to estrogen have been established as risk factors, including early menarche, late menopause, and obesity in postmenopausal women. Factors that reduce the cumulative exposure to estrogens, such as early first pregnancy, multiple full-term pregnancies, oophorectomy in premenopausal women, and physical activity, are associated with a reduced risk of breast cancer. Consistent with these observations, increased serum levels of estrogens are associated with postmenopausal breast cancer (Table 20–1). Use of hormone replacement therapies in menopausal women, particularly those that combine an estrogen and a progestin, also increase the risk of breast cancer (Banks et al, 2008).
TABLE 20–1 Hormonally related epidemiological risk factors for breast cancer.
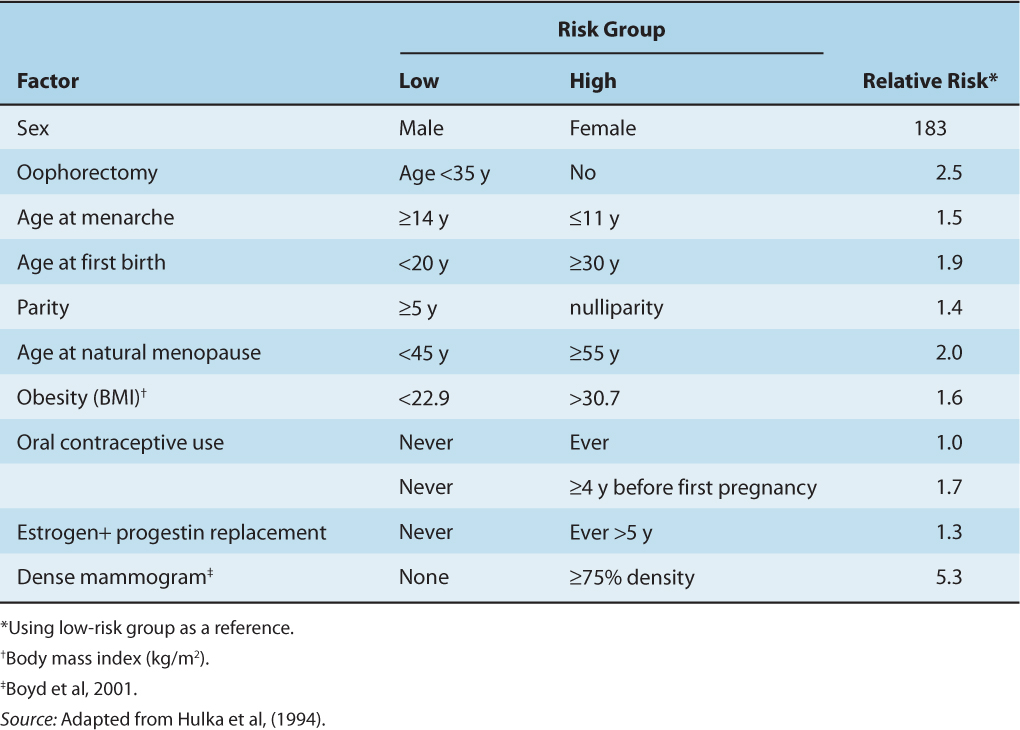
Increased ER expression in normal breast epithelium is a risk factor for breast cancer (Khan et al, 1998), as is increased mammographic breast density (Martin et al, 2009) and increased circulating IGF-I levels (Key et al, 2010). These latter 2 factors are correlated and may be associated functionally (Becker and Kaaks, 2009). Estrogens have been shown to increase, and tamoxifen to decrease, mammographic density, and the intimate association and crosstalk of the IGF-I signaling pathway with the ER signaling pathway in target cells may also play a role in the risk of breast cancer (Key et al, 2010).
Women with a family history of breast cancer are at increased risk of breast cancer and 2 genes, BRCA1 and BRCA2, when carrying a germline mutation, are associated with an inherited predisposition to breast cancer (see Chap. 5, Sec. 5.3.4 and Chap. 7, Sec. 7.6.3). Only 5% of all breast cancer incidence can be attributed to inherited mutations in these genes and less than 10% of all breast cancer incidence can be attributed to an inherited predisposition. The importance of environmental factors is highlighted by the observation that the risk of breast cancer in Asian women rises over a few generations following migration to Western countries (Ziegler et al, 1993). Most breast cancers are sporadic, although polymorphisms in genes associated with the biosynthesis of estrogens (Thompson and Ambrosone, 2000) and factors that regulate ER activity such as AR (Giguere et al, 2001), may influence breast cancer development. Breast cancer risk is complex, probably because of the influence of multiple genes with the environment.
The primary role of estrogen in breast cancer is thought to be because of its proliferative effect on breast epithelium. A complex interplay of steroid hormones, growth factors, extracellular matrix, and their respective receptors is likely involved. Genotoxic effects of steroids cannot be excluded, although the relative roles of the different mechanisms remain unclear (Lin et al, 2009; Pauklin et al, 2009).
20.3.2 Development of Breast Cancer
Some of the cellular events associated with the natural history of breast cancer are illustrated in Figure 20–8A. Most invasive breast cancers arise from the epithelial cells of the terminal duct lobular unit (TDLU). Histopathological studies have identified a series of premalignant breast lesions referred to as hyperplasia without atypia, usual ductal hyperplasia (UDH), atypical hyperplasia (AH), and ductal carcinoma in situ (DCIS). These lesions are associated with increasing risks of developing invasive breast cancer. For example, AH is associated with a 5-fold increased risk, and DCIS is associated with a 10-fold increased risk (Page et al, 2000). Comparisons between premalignant and/or preinvasive lesions in the same biopsy sample as invasive breast cancers have identified common genetic abnormalities, suggesting that the malignant lesions are clonally derived from the earlier lesions (Allred and Mohsin, 2000; Gong et al, 2001).
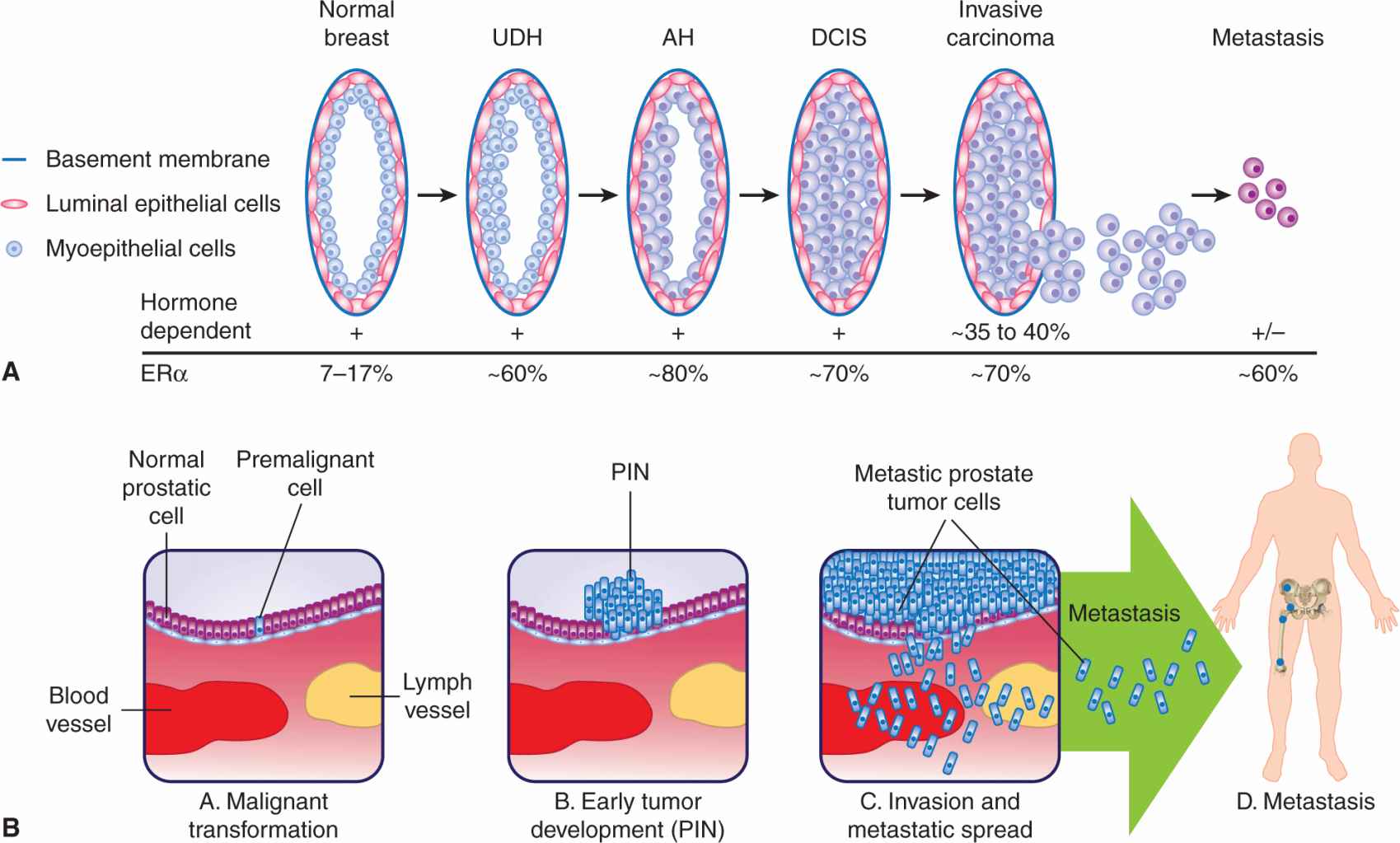
FIGURE 20–8 An overview of the natural history of cancers of the breast and the prostate. A) Normal breast epithelium can undergo a stepwise transition from a series of premalignant breast lesions referred to as hyperplasia without atypia/usual ductal hyperplasia (UDH), atypical hyperplasia (AH), and ductal carcinoma in situ (DCIS) leading to invasive carcinoma and metastasis (adapted from Myal et al, 2010). The relative hormonal dependency and receptor status at the various stages are indicated. B) A normal prostate epithelial cell can develop into a premalignant tumor cell that can give rise to prostatic intraepithelial neoplasia (PIN), and then become an invasive carcinoma.
Although normal development of the mammary gland is dependent on the presence of ERα and only a rudimentary ductal remnant is present in “knockout” mice that do not have the ERα gene (Korach, 1994), only 7% to 17% of normal breast epithelial cells express ERα (Clarke et al, 1997). In contrast more than 70% of human breast tumors are ERα+, and often the level of ERα expression in tumor cells is higher than that found in normal breast luminal epithelial cells. Most hyperplastic lesions, with or without atypia, show increased expression of ERα and increased frequency of ER+ cells compared to normal epithelium, and more than 70% of DCIS are ERα+, similar to invasive breast cancer (Allred and Mohsin, 2000). In normal breast epithelial cells, ERα and Ki67, a marker of cell proliferation, are rarely, if ever, coexpressed (Anderson et al, 1998), suggesting that either ERα-expressing cells are incapable of proliferating or that ERα must be downregulated before normal breast epithelial cells can proliferate. This inverse relationship is maintained in UDH, but is lost in AH, DCIS, and ERα+ invasive breast cancer cells (Shoker et al, 1999). Thus an alteration in estrogen responsiveness and/or mechanism of estrogen action occurs during the development of breast cancer.
The second ER, ERβ, is also expressed in both normal and neoplastic human breast tissues (Leygue et al, 1998; Roger et al, 2001). Unlike ERα, ERβ does not play a pivotal role in development of the mammary gland as “knockout” mice for ERβ have normal development of the mammary gland (Couse and Korach, 1999). Expression of ERβ is much more frequent than ERα in normal human and rodent mammary glands, and its expression generally declines during breast cancer development (Leygue et al, 1998; Roger et al, 2001). Altered expression of several coregulators of ERα occurs during breast tumori-genesis (Murphy et al, 2000; Gojis et al, 2010) with a general trend toward increased expression of known coactivators and decreased expression of known corepressors; this observation suggests that a marked upregulation of ERα signaling occurs during breast tumorigenesis.
Growth suppression pathways associated with transforming growth factor (TGF)-β (see Chap. 8, Sec. 8.4.4) are altered in some early lesions. For example TGF-β receptor type II is highly expressed in normal breast epithelium, but is downregulated in some UDH lesions and identifies a group of women at higher risk of developing breast cancer (Gobbi et al, 1999). Altered growth factor pathways that stimulate the cell cycle, for example overexpression of human epidermal growth receptor 2 (HER2) (neu/erbB-2) and cyclin D1, and/or inactivation of tumor-suppressor genes such as p53, can be detected in some premalignant lesions such as DCIS (Allred and Mohsin, 2000; see Chap. 7, Secs. 7.5.3 and 7.6.1).
Breast cancer is extremely heterogeneous, as defined by the great variability that is seen in morphology, gene expression patterns, and behavior of individual tumor cells within any given tumor (Campbell and Polyak, 2007). The origin of this heterogeneity and the target cell(s) of origin of breast cancer are unknown, but 2 hypotheses that are not mutually exclusive have been suggested: the cancer stem or initiating cell and the clonal evolution hypotheses (see Chap. 13, Sec. 13.2). Substantial progress in defining the normal human mammary stem cell and the mammary epithelial hierarchy has been made at the molecular and functional levels (Eirew et al, 2008; Visvader, 2009). Despite the importance of ERα signaling in human breast cancer and in the normal development of the mammary gland, normal mammary stem cells and/or populations enriched for stem cell repopulating characteristics, appear not to express ERα or PR, supporting the idea that at least in the normal mammary gland the effect of steroid hormones on proliferation is indirect. Expression of ERα seems to appear within the mammary epithelial hierarchy at the committed luminal progenitor stage (Visvader, 2009) and the expression of PR may occur earlier than this stage in the common-bipotent epithelial progenitor cells (Raouf et al, 2008). Support for the existence of a population of human breast cancer cells with cancer initiating/stem cell-like characteristics is emerging (Al-Hajj et al, 2003; Eirew et al, 2008; Lim et al, 2009; Korkaya et al, 2011; see Chap. 13, Sec. 13.4.5), but whether the target cell for breast cancer is the normal mammary stem cell or other cells along the mammary epithelial hierarchy is unknown. The use of molecular signatures from molecular expression analysis is helping to resolve such issues, as there are now data linking newly identified breast cancer subtypes (Fig. 20–9, discussed below) with their closest normal mammary epithelial counterpart based on similarities of gene expression profiles (Visvader, 2009). However, it is also possible that cancer-initiating cells could be derived from mutation of tissue-specific progenitors or more differentiated cells which, because of mutation, acquire self-renewal capacity (Hershkowitz, 2010).
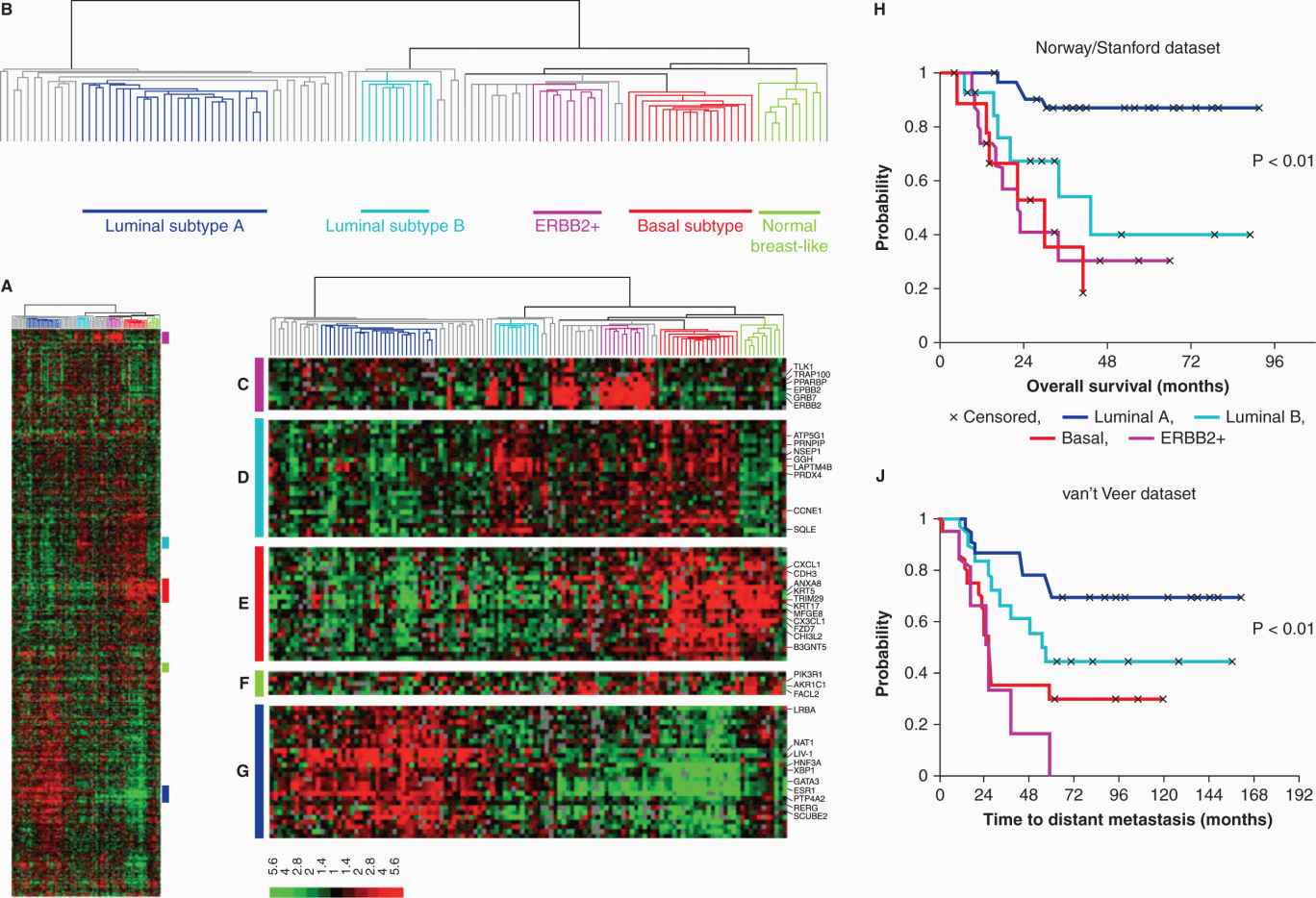
FIGURE 20–9 Molecular subtypes of breast tumors. A) Gene expression microarray analyses where expression of more than 500 genes, at the RNA level, has been measured in 122 different breast tissue samples (115 tumors, 7 non-malignant). Each column represents one breast sample, and each row represents the expression level of one gene. The scale bar represents the fold-change for any given gene relative to the median level of expression of all samples. Green is decreased and red is increased expression. B) Dendrogram of the heirarchical clustering of the tissues with respect to similarities in their gene expression patterns, into 5 subgroups shown by the different colors (dark blue, luminal A; aqua-blue, luminal B; purple, ERBB2+; orange, basal like; green, normal breast-like). Tumors with low correlations to these 5 subtypes are shown in gray. C) Gene cluster showing ERBB2 oncogene and co-expressed genes. D) Gene cluster associated with luminal subtype B. E) Gene cluster associated with the basal subtype. F) Gene cluster relevant for the normal breast-like group. G) Cluster of genes including the estrogen receptor (ESR1) highly expressed in luminal subtype A tumors. This classification is associated with different clinical outcomes as illustrated by Kaplan-Meier analyses from 2 different datasets shown in H and J where the colors of the survival curves correspond to those of the molecular subtypes. H) Overall survival for 72 patients with locally advanced breast cancer. J) Time to development of distal recurrence in 97 patients in another study (Adapted from Sorlie et al, 2003).
20.3.3 Progression of Invasive Breast Cancer
Amplification and upregulation of expression of several oncogenes, including those encoding growth factors such as EGF, TGF-α, IGF, and their receptor tyrosine kinases, for example, EGFR, HER2 (neu/erbB-2), and IGFR, together with inactivation or downregulation of tumor-suppressor genes, such as p53, Rb, or BRCA1, have been associated with breast cancer progression. The introduction of comparative genomic hybridization (CGH), fluorescence in situ hybridization (FISH), spectral karyotyping (SKY), DNA microarrays, and next-generation sequencing (Shah et al, 2009; see Chap. 2) is allowing a more global analysis of the molecular genetic alterations that occur during cancer progression.
Prognosis of patients with invasive breast cancer is related to lymph node involvement, tumor size, histological grade, and steroid receptor status. Approximately 70% of all primary invasive breast tumors are ER+, and in general, these tumors are more differentiated, less aggressive, and have lower levels of growth factor receptors compared to ER– tumors. In contrast, consistent evidence has accumulated to show that young patients (younger than age 40 years) with ER+ tumors have a worse prognosis than those with ER– tumors (Aebi, 2005).
Not only is ER status a prognostic factor, but it also is a marker of response to treatment, as is discussed subsequently. Until recently, the only molecular classifiers of breast cancers were ERα, PR, and HER2/ErbB2. ERα and overexpressed/amplified HER2 are the targets of endocrine therapies and the humanized antibody trastuzumab, respectively. HER2/ErbB2 is amplified and/or overexpressed in up to 20% of all breast tumors, and these tumors are aggressive with generally poor outcome, but the development of trastuzumab targeted to HER2 overexpressed protein has improved the outcome for these patients (Di Cosimo and Baselga, 2010). The advent of global molecular profiling technologies has led to a new way of classifying human breast cancer and is the basis of a more individualized approach to breast cancer prognostication and therapy. Most effort has focused on classification according to gene-expression profiling at the mRNA levels using microarray chip technology (Perou et al, 2000). However, a combination of transcriptomic, epigenetic, proteomic, and genomic analyses may be necessary to appreciate the true heterogeneity of breast cancer (Weigelt and Reis-Filho, 2009).
Despite the limitations outlined above, gene expression analysis has resulted in human breast tumors being classified into 5 different subtypes, with potentially a sixth recently identified (Visvader, 2009). The subtypes are called luminal A, luminal B, HER2 overexpressing, basal-like, normal breast-like (Sorlie, 2004) and, most recently, a claudin-low subtype has been described (Creighton et al, 2010). The different subtypes are clinically relevant as some are associated with different clinical outcomes (see Fig. 20–9). ERα and PR are only expressed in the luminal A and B subtypes, although about half of the HER2 overexpressing subtype also can express ERα. Furthermore, although alternative hypotheses cannot be excluded (Stingl and Caldas, 2007), the similarity of expression signatures to those represented in the mammary epithelial hierarchy may provide information concerning the cell of origin of the subtypes and identify the nature of the cancer-initiating cells such that new treatments can be designed to target them. However, these molecular classifications are research tools and are not used routinely in clinical practice, in contrast to the determination of ER, PR, and HER2 expression.
The triple negative (ERα-negative, PR-negative, and HER2-negative) group of tumors represents a challenge in breast cancer because of their lack of responsiveness to endocrine therapies and trastuzumab. These tumors are aggressive with increased risk of metastasis, but in some cases respond well to chemotherapy. The triple negative or basal-like subtype is itself heterogeneous, and a better understanding of the molecular heterogeneity of this subgroup is necessary to improve clinical outcome (Di Cosimo and Baselga, 2010). Most breast cancers arising from BRCA1 mutation carriers are triple negative (Podo et al, 2010), whereas most breast cancers arising from BRCA2 mutation carriers are ER+ and have a luminal molecular profile (Bane et al, 2007).
A proportion of all tumor subgroups, despite originally responding to treatment, will recur and progress, having acquired resistance to targeted therapies. Most ER+ tumors treated with endocrine therapies develop resistance despite the continued expression of ERα. Mechanisms of resistance to endocrine therapies are discussed below, but a challenge is to identify biomarkers that predict early resistance to endocrine therapy so that either more aggressive therapy can be used sooner, or a rational combination of therapies can be given. The use of new technologies that allow a more global analysis of the molecular and genetic alterations occurring in tumors holds promise with respect to achieving this goal (Wood et al, 2007).
20.3.4 Risk Factors for Prostate Cancer
Carcinogenesis of the prostate involves genetic and environmental influences with no obvious etiological agent. Risk factors include family history, age, and race (Hsing and Chokkalingam, 2006). First-degree male relatives of prostate cancer patients have an approximately 2.5-fold increase in risk, and the risk of prostate cancer appears to be higher for relatives of women with breast cancer. Men carrying a germline BRCA2 mutation have increased risk of developing a more aggressive prostate cancer by 3.2-fold compared with noncarriers (Gallagher et al, 2010). However, hereditary factors most commonly affect men with early onset disease and are responsible for relatively few cases (<10%). Diet is probably important, with the predominantly vegetarian diet of Asians providing a protective influence, whereas the high intake of red meat associated with a typical American diet is likely related to increased risk (Denis et al, 1999). Epidemiological studies have linked obesity with a range of cancer types, although its role in the development and progression of prostate cancer has not been elucidated. The association of obesity with numerous hormonal changes that influence endocrine pathways may contribute to prostate cancer development and progression (Calle and Kaaks, 2004).
Prostate cancer is a disease of the elderly, with more than 75% of cancers diagnosed in men older than 65 years of age. However, microfoci of high-grade prostatic intraepithelial neoplasia (PIN), the presumed precursor of the disease, can be found in men in their third and fourth decade of life (see Fig. 20–8B). Most of the early tumors are microscopic, generally well to moderately differentiated, and tend to be multifocal. The frequency with which these neoplastic lesions are seen in autopsy material is similar among African Americans, white Americans, and Japanese men, but the incidence of clinical disease is higher in African American men and lower in Japanese men. In Japanese immigrants, the incidence of prostate cancer rises to levels near those of white Americans within 2 generations, suggesting the involvement of environmental factors. Collectively these observations suggest that the critical event in the natural history of prostate cancer is tumor promotion rather than tumor initiation, and that promotion and progression of this cancer are strongly influenced by epigenetic or adaptive processes.
20.3.5 Development of Prostate Cancer
Two pathological conditions that frequently coexist with latent and clinical prostate cancer are benign prostatic hyperplasia (BPH) and PIN. BPH shares many biological properties with prostate cancer, including androgen regulation of growth and increasing prevalence with advancing age. However, BPH is neither a premalignant lesion nor a precursor of invasive prostate cancer. A more likely candidate for this role is PIN, which is characterized by cytological atypia of proliferating luminal epithelium within preexisting acini and ducts with no penetration of the basement membrane (see Fig. 20–8B). Histologically, PIN is generally subdivided into low or high grade and autopsy studies reveal that high-grade PIN is found in association with cancer in 60% to 95% of malignant prostates and that a wide spectrum of molecular/genetic abnormalities are common to both high grade PIN and prostate cancer (Sakr and Partin, 2001). Specific chromosomal alterations (eg, loss of 8p, 10q, 16q, 18q, and gain of 7q31, 8q), amplification of the c-myc gene, along with changes in telomerase activity, cell-cycle status, and proliferative indices, suggest, collectively, that high-grade PIN is intermediate between benign epithelium and prostatic carcinoma (Sakr and Partin, 2001). A related staining profile for growth factors and for the AR has been demonstrated in the luminal epithelium of high-grade PIN and in carcinoma, with a tendency to higher expression of membrane EGFR, c-erbB-2, and cytoplasmic TGF-α, and lower levels of fibroblast growth factor (FGF)-2, than in glands with low-grade PIN or BPH (Harper et al, 1998). In addition to being a precursor for prostate cancer, it is likely that PIN predates invasive cancer by at least a decade and thus may serve as a predictive marker for the disease.
Although uncertain, prostate and other cancers may arise as a consequence of genetic alterations in a stem cell population (Lawson and Witte, 2007). Indeed, their longevity makes stem cells more likely to develop genetic alterations over time that may eventually culminate in cancer. Moreover, unlike mature prostate cells, primitive stem cells can thrive under androgen depletion, and it has been shown that these cells can subsequently regenerate prostate tissues with androgen stimulation. These castration-resistant cells were found originally to be of basal origin (Mulholland et al, 2009); however, the recent finding in mice of a luminal stem cell population that displays castration-resistant characteristics during prostate regeneration (Wang et al, 2009) suggests that there may be at least 2 different cell types that play a role in the development of castration-resistant phenotypes. However, only a small proportion of cancer cells within a tumor may possess stem cell properties (see Chap. 13, Sec. 13.4). These findings may be of critical importance to identify novel therapeutic approaches for future clinical management.
20.3.6 Progression of Invasive Prostate Cancer
When organ confined, prostate cancer is potentially curable by prostatectomy or radiation therapy. The problem here is selecting patients who need such treatment, as many will never develop symptomatic disease, whereas others will have occult metastases. In men with locally advanced or metastatic disease, treatment is largely palliative with androgen withdrawal as first-line treatment. The clinical approaches used for androgen withdrawal are described below. Despite high initial response rates on the order of 80%, patients will inevitably progress to hormone-independent disease in a manner analogous to that seen following hormonal therapy for metastatic breast cancer. Response to androgen ablation therapies depends on the degree of retention by the tumor of the capacity for activation of apoptosis after androgen withdrawal. However, many so-called hormone-independent cancers may in fact still be dependent on androgens, but resistant to medical or surgical castration, hence the term “castration-resistant” prostate cancer (CRPC).
20.4 HORMONAL THERAPIES FOR BREAST AND PROSTATE CANCERS
20.4.1 Breast Cancer
Endocrine therapies for patients with breast cancer are aimed at inhibiting the proliferative effect of estrogen on breast cancer cells. As outlined in Figure 20–2A, this aim is generally achieved in 2 ways: decreasing the level of circulating and/or local estrogen, or blocking the action of estrogen on the target tissue. Only those breast tumors that express ER benefit from endocrine therapies.
Reduced levels of estrogen can be achieved surgically, by oophorectomy, and pharmacologically. In premenopausal women, options are surgical or radiation-induced oophorectomy, or the use of LHRH agonists, which initially stimulate and then block the LHRH receptor in the pituitary gland. This approach leads to a reduction in levels of gonadotrophins, which stimulate the ovary to synthesize estrogens.
In postmenopausal women, residual levels of estrogen can be reduced by selective aromatase inhibitors that inhibit the conversion of weak androgens to estrogens (see Fig. 20–1). As shown in Figure 20–10, aromatase inhibitors fall into 2 main classes—the steroidal (eg, exemestane) and the nonsteroidal (eg, anastrozole and letrozole). The 2 classes differ in their mechanism of action. The steroidal inhibitors compete with endogenous substrates for the active site of the enzyme, and are processed to intermediates that bind irreversibly to the active site, causing inhibition. The nonsteroidal inhibitors also compete with the endogenous substrates for the active site, but form a strong, although reversible, coordinate bond with the heme iron atom, excluding endogenous substrates and oxygen from the enzyme. Removal of the nonsteroidal inhibitor results in reversal of enzyme inhibition. These agents are alternatives to tamoxifen (see below) as first-line treatment of postmenopausal women. Aromatase inhibitors are not effective alone in premenopausal women with ER+ breast cancer because the aromatase inhibitor-induced reduction in plasma estrogen increases follicle-stimulating hormone (FSH), which can override the block as a result of inhibition of aromatase, and eventually lead to increased estrogen from the ovary. Use of aromatase inhibitors, after oophorectomy or LHRH agonists have induced menopause, in premenopausal women is being evaluated (Di Cosimo and Baselga, 2010). Progestins, such as megestrol acetate, are useful as third-line endocrine therapies in breast cancer, and part of their mechanism of action could include the increased expression and/or activity of enzymes, which can metabolize strong estrogens into weaker compounds.
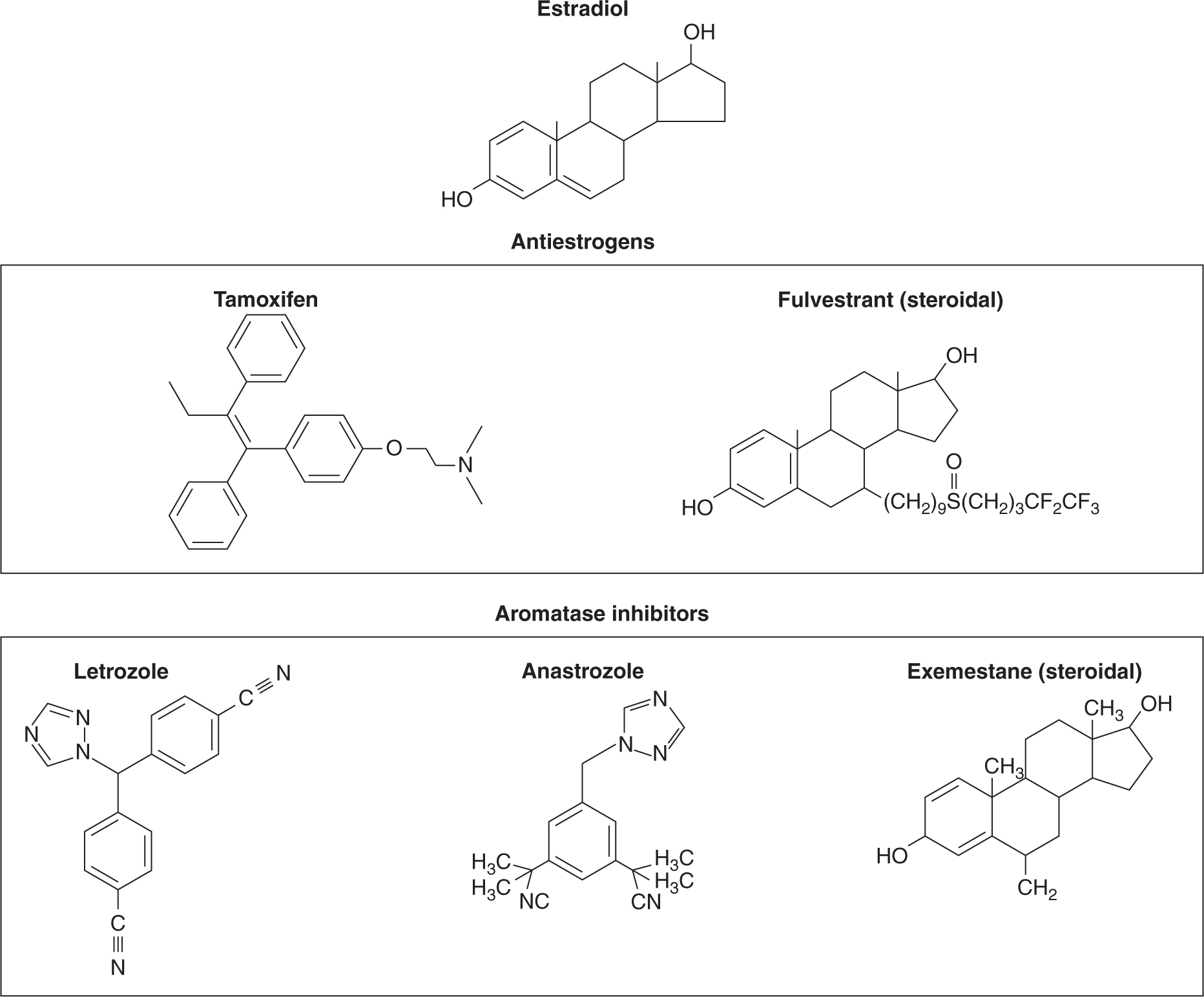
FIGURE 20–10 Structures of antiestrogens and aromatase inhibitors. The antiestrogens block estrogen binding to its receptor and the steroidal forms also increase turnover of the receptor protein. The aromatase inhibitors block the formation of estrogens from androgen precursors.
The nonsteroidal antiestrogen, tamoxifen has been the agent of choice for first-line endocrine therapy for the treatment of ER-positive and/or PR-positive breast cancer (see Fig. 20–10). It leads to improved survival when used as adjuvant therapy for women with receptor-positive breast cancer of all ages, and may decrease the incidence of breast cancer in groups of women at high risk for the disease. Tamoxifen and its more active metabolites, 4-hydroxytamoxifen and 4-hydroxy-desmethytamoxifen (endoxifen), competitively inhibit the binding of estradiol to the ligand-binding site of the ER in a dose-dependent manner (see Fig. 20–3). Because tamoxifen requires metabolism to more active metabolites, alterations in the activity of the enzymes responsible for its metabolism (Fig. 20–11) may affect an individual patient’s responsiveness to tamoxifen. This possibility is discussed in more detail later.
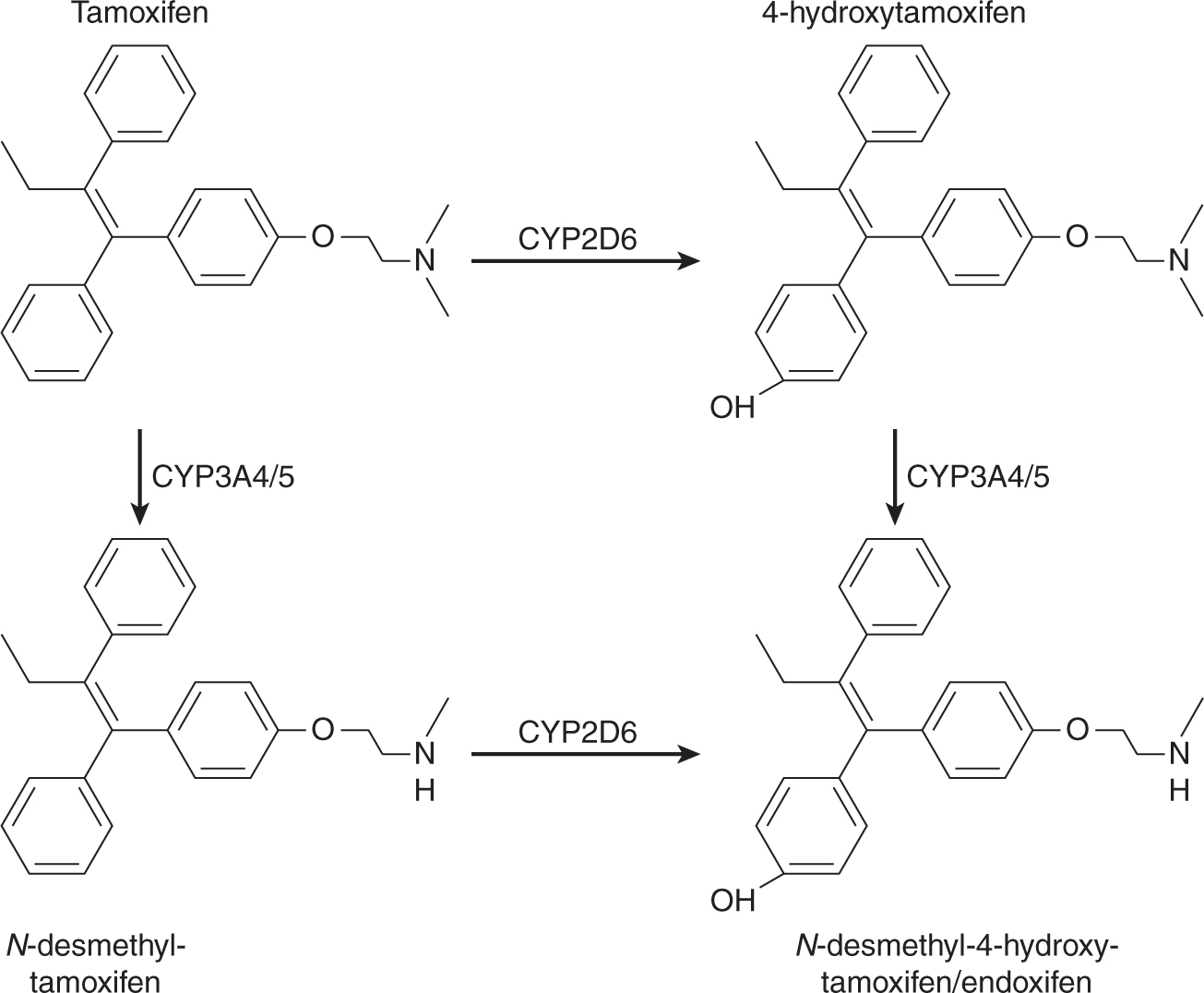
FIGURE 20–11 Metabolism of tamoxifen to more active metabolites such as 4-hydroxytamoxifen and N-desmethy-4-hydroxytamoxifen (endoxifen).
When tamoxifen or similar compounds bind to the ER, they result in conformational changes that allow the inactive receptor to bind to DNA but not to activate transcription (see Fig. 20–4). X-ray crystallography studies of the ER ligand domain bound to either estradiol or 4-hydroxytamoxifen, show that estradiol binding causes formation of a hydrophobic cleft on the surface of this domain which serves as a docking site for coactivators. In contrast, antiestrogens displace part of the receptor, blocking this site, and therefore blocking coactivator access (Shiau et al, 1998). Compounds like tamoxifen have both estrogenic and antiestrogenic properties, depending on the cell type or the promoter of any particular target gene. Tamoxifen has estrogenic effects in bone and in cardiovascular tissue (which are desirable to prevent osteoporosis and cardiac disease) and in the uterus (which is undesirable, as it may stimulate proliferation and lead to a low incidence of uterine cancer). Compounds with different selectivity in their estrogenic and antiestrogenic properties are referred to collectively as selective estrogen receptor modulators (SERMs). The basis of their differences is in the slightly different conformational changes that they induce in the ER, leading to differential abilities to interact with a variety of coregulators, whose expression and/or activation will vary between cell types and tissues, as illustrated in Figure 20–12 (McKenna and O’Malley, 2000). Because breast tumors often develop resistance to tamoxifen without loss of ER expression, and because tamoxifen has undesirable estrogenic properties in the uterus, other SERMs are being investigated for their usefulness in breast cancer treatment and prevention. For example, raloxifene is a SERM that has estrogenic effects on bone and the cardiovascular system, but is antiestrogenic in the breast and uterus (O’Regan and Jordan, 2001). A clinical trial (Study of Tamoxifen and Raloxifene [STAR]) has found it to have comparable efficacy to tamoxifen in breast cancer prevention in high risk women, without increasing the risk of endometrial cancer (Vogel et al, 2006).
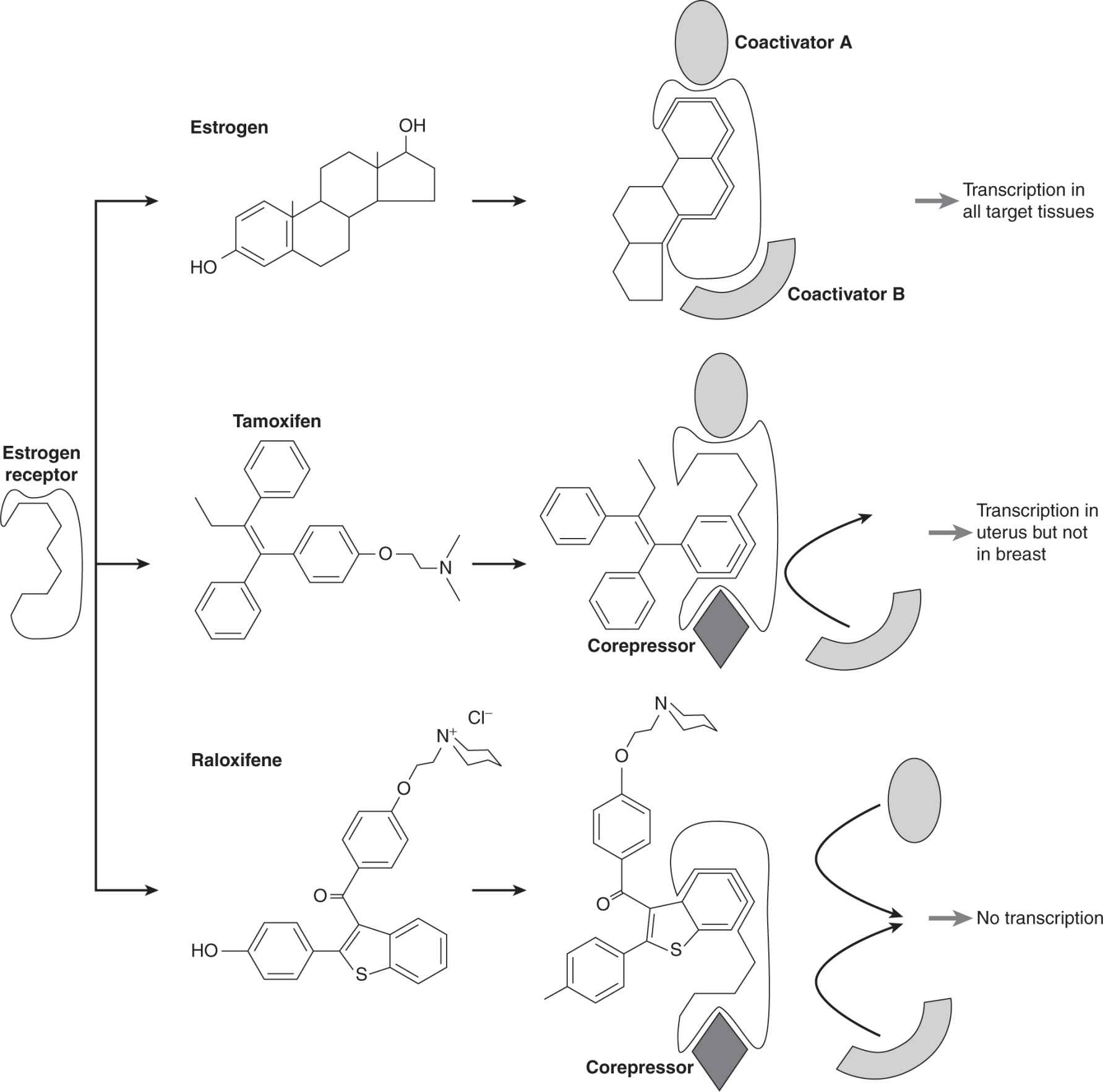
FIGURE 20–12 Schematic of conformational changes induced by different SERMs (estrogen, tamoxifen, and raloxifene) resulting in differential recruitment of coregulators and differential activity. Estrogen binds to the estrogen receptor (ER) causing a conformational change that leads to docking of appropriate coactivators (hypothetical coactivator A and coactivator B) with ER in all target tissues and resulting in enhanced transcription of target genes. Tamoxifen binds to ER giving rise to a different conformation such that it docks coactivator A and a hypothetical corepressor that in one tissue (breast) inhibits estrogen action, but in another tissue partially activates estrogen action (uterus). Another SERM called raloxifene binds to ER, resulting in a different conformation such that it only docks the corepressor and inhibits estrogen action in both target tissues.
Steroidal antiestrogens, such as fulvestrant (see Fig. 20–10), that show little if any estrogenic activity (in particular in the uterus) retain activity in some women whose breast cancers have acquired resistance to tamoxifen. The mechanism of action of these “pure” antiestrogens is distinct from the partial antiestrogens such as tamoxifen, in that they downregulate ER expression by increasing its degradation, as well as inactivate the ER complex (O’Regan and Jordan, 2001). Such compounds have been named selective estrogen receptor downregulators (SERDs). Because of its steroidal nature, fulvestrant is not suitable for oral administration and is administered by intramuscular injection, which is a disadvantage. High-dose progestin treatment that is used as a third-line endocrine treatment may also act partially by downregulating expression of the ER in breast tumors.
20.4.2 Hormonal Therapy for Different Stages of Breast Cancer
Systemic adjuvant therapy given after surgical removal of the primary tumor is aimed at eliminating subclinical, micrometastatic cancer cell deposits that have spread from the original tumor site. Adjuvant hormonal therapies, such as tamoxifen or ovarian ablation in premenopausal women and tamoxifen or aromatase inhibitors (given for 5 years) in postmenopausal women with breast cancer, have been shown to increase both relapse-free and overall survival of women with ER+ early breast cancer, when used either alone, or following adjuvant chemotherapy (Early Breast Cancer Trialists’ Collaborative Group, 2005). The hormonal therapies are also used to treat metastatic breast cancer, which is not curable, and the goals of therapy are to prolong survival and to maximize the quality of life of the patient. The hormonal treatments are often used in sequence, and women who respond to initial endocrine treatment have approximately a 50% chance of responding to a second agent, while other hormonal agents, including megestrol acetate and fulvestrant, may lead to further responses when used as third-line treatment in selected patients. Despite the responsiveness of advanced ER+ breast cancer to initial endocrine therapy, tumors eventually develop resistance to all forms of endocrine therapy. The multiple mechanisms thought to be involved in resistance to hormonal therapy are explored in Section 20.5.
Neoadjuvant (preoperative) endocrine therapy (with or without chemotherapy) is also used in some patients to reduce the size of the primary breast cancer, thereby increasing the possibility of breast-conserving surgery and allowing assessment of response to treatment in primary tumors.
20.4.3 New Approaches to Hormonal Treatment of Breast Cancer
Genomic molecular profiles (mutation and expression analyses) of human breast tumors can identify key activated pathways in ER+ tumors to provide a rational basis for using combinations of therapies in the most appropriate patients (Johnston, 2010). Several clinical trials of combinations of endocrine therapies with targeted biological agents are ongoing. The use of pharmacogenetics (see Chap. 18, Sec. 18.1.3) to identify those patients more likely to have adverse side effects or less likely to benefit from a particular treatment or combination of treatments is another important focus in cancer therapy. A more precise understanding of the mechanisms responsible for resistance to different endocrine therapies, may also lead to changes in sequencing and scheduling of treatment. For example, a potential mechanism for the development of resistance to aromatase inhibitors is upregulation of growth factor and survival pathways, and this resistance might be reversed by periods of time off treatment, in contrast to continuous treatment.
20.4.4 Prostate Cancer
Approximately 80% of patients with metastatic prostate cancer will respond to androgen withdrawal therapy, as indicated by a fall in the serum marker of the disease, PSA, and by an improvement in symptoms (most often pain caused by metastases in bone). Such treatment is associated with a median progression-free survival of 1 to 2 years and overall survival of 2 to 4 years. The failure to eradicate the entire malignant population by androgen withdrawal results in progression to castration resistance, as manifested by a rising serum PSA and/or clinical signs and symptoms. The goal of any form of androgen withdrawal therapy is to activate apoptosis or block cell proliferation in prostate tumor cells by inhibiting androgen-dependent signaling (see Fig. 20–2B). There are several methods for achieving androgen withdrawal, either by interfering with the synthesis or metabolic conversion of androgens or with their ability to interact with the AR. However, as most castration-resistant prostate tumors have normal or elevated levels of AR, there is no prognostic value in measuring these receptors.
Bilateral orchiectomy (castration) is the most direct way to block androgen stimulation of the prostate and has the advantages of low morbidity, low cost, and high compliance. However, the associated psychological problems have decreased its practice in favor of medical castration using LHRH agonists. LHRH agonists include goserelin, leuprolide, and buserelin, which can be administered as long-acting (3 to 4 months) formulations to block the secretion of LH by the pituitary gland and thereby inhibit the synthesis of testosterone by the testis (see Fig. 20–2B). Initially, LHRH agonists cause a rise in LH and testosterone that is termed a “flare” response; this effect is avoided by temporary coadministration of an antiandrogen to inhibit AR signaling in the tumor cell. A rising serum PSA level is usually the earliest manifestation of progression following initial endocrine therapy, often predating clinical evidence of progression by more than 6 months.
Antiandrogens, some of which are shown in Figure 20–13, act competitively with testicular or adrenal androgens to block AR activation within the prostate cell (see Fig. 20–3) and apart from transient use to prevent the flare associated with LHRH agonists, are usually added as second-line hormonal therapy at time of progression. Nonsteroidal antiandrogens, such as flutamide or bicalutamide (Fig. 20–13), have no gonado-tropic or hypothalamic feedback activity to suppress circulating levels of testosterone, whereas steroidal antiandrogens such as cyproterone acetate do possess this feedback activity (see Fig. 20–2B). Approximately 30% of patients who have responded and then progressed after initial hormone therapy will respond to the subsequent addition of an antiandrogen. At the time of progression following this treatment, discontinuation of the antiandrogen leads to further response in approximately 20% of patients. The underlying molecular mechanism responsible for the antiandrogen withdrawal syndrome is not fully understood, but may relate to altered AR ligand specificity. A new, more potent nonsteroidal antiandrogen, called enzalutamide (MDV3100), has been developed. Enzalutamide binds the AR with greater affinity than bicalutamide, impairs its nuclear translocation, and may have negative effects on the binding of AR to androgen response elements and the recruitment of coactivators. The results of Phase II trials showed that enzalutamide had substantial antitumor effect in men who had progressed during or after treatment with first-generation antiandrogens, with few side effects (Tran et al, 2009; Scher et al, 2010). A randomized Phase III trial has shown that enzautamide improves the survival of men with castrate-resistant prostate cancer, whose disease has progressed after receiving chemotherapy (Scher et al, 2012).
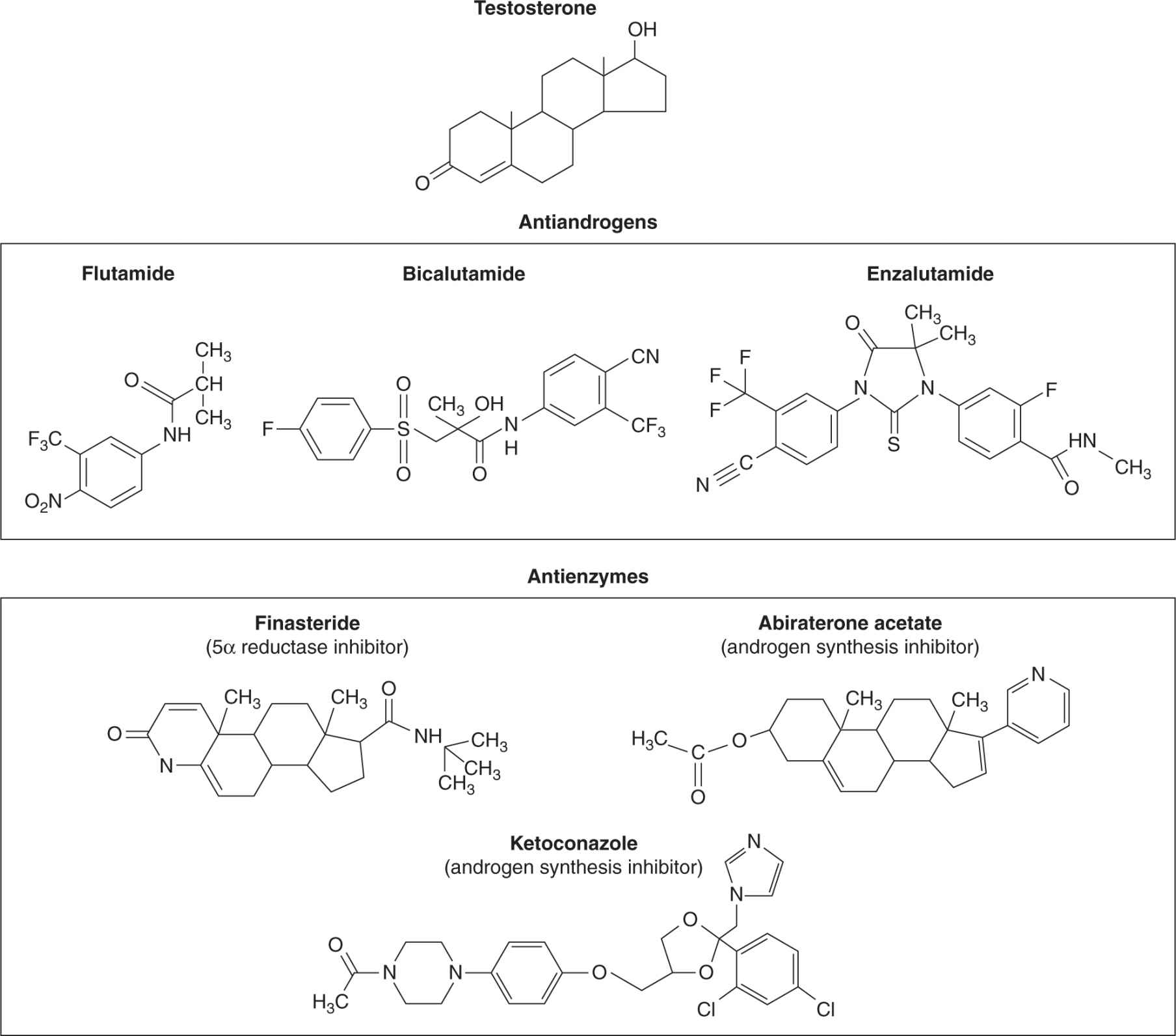
FIGURE 20–13 Structures of antiandrogens and antienzymes that block androgen metabolism. The antiandrogens block androgen binding to its receptor. The antienzymes inhibit the conversion of testosterone to the more active form dihydrotestosterone (5a reductase inhibitors) or block androgen synthesis (ketoconazole and abiraterone acetate).
Other second- or third-line strategies include the use of ketoconazole to inhibit steroid synthesis in general, although this drug is likely to be replaced by abiraterone acetate, which is a more specific and potent inhibitor of androgen synthesis (see Fig. 20–13, see also Sec. 20.5.3). Abiraterone acetate has shown a substantial improvement in time to disease progression of men with metastatic castrate-resistant prostate cancer, and improved survival when used to treat men with late-stage metastatic prostate cancer, who had progressed after receiving chemotherapy (de Bono et al, 2011; Ryan et al, 2012). Glucocorticoids, such as prednisone and hydrocortisone, can inhibit production of adrenocorticotropic hormone (ACTH), which leads to decreased production of weak androgens by the adrenal gland, and can also lead to tertiary responses (see Fig. 20–2B). Estrogens have also been used to suppress LH and FSH by feedback effects on the pituitary; they have rare therapeutic effects in patients who have already received several hormonal treatments and may have direct effects on prostate cancer cells, which express both ERα and ERβ. Irrespective of the type of androgen withdrawal treatment used, the side effects can include hot flushes, loss of libido and sexual potency, gynecomastia, lethargy, depression, loss of bone and muscle mass, and metabolic syndrome with increased diabetes and cardiac events.
Many patients have received treatment with “complete androgen blockade,” which combines orchiectomy or an LHRH agonist with a nonsteroidal antiandrogen. Although a metaanalysis of several trials comparing complete androgen blockade with conventional medical or surgical castration showed no significant survival advantage (Prostate Cancer Trialists’ Collaborative Group, 2000), the advent of new more potent antiandrogens, such as enzalutamide, may help to improve survival.
20.4.5 Intermittent Hormonal Therapy for Prostate Cancer
In addition to acting as mitogens to induce DNA synthesis and cell proliferation, estrogens and androgens are potent differentiating agents. In studies with castrated rodents, administration of small amounts of androgen was shown to induce markers of differentiation in the prostate without stimulating rounds of cell proliferation. This conditioning effect of androgens on surviving cells allowed them to retain desirable, hormone-regulated traits of differentiation, and the capacity to undergo apoptosis upon hormone withdrawal. Hence periodic exposure to hormones might maintain hormone responsiveness of prostate tumors. Figure 20–14 shows an idealized representation of how intermittent hormone suppression might regulate tumor growth.
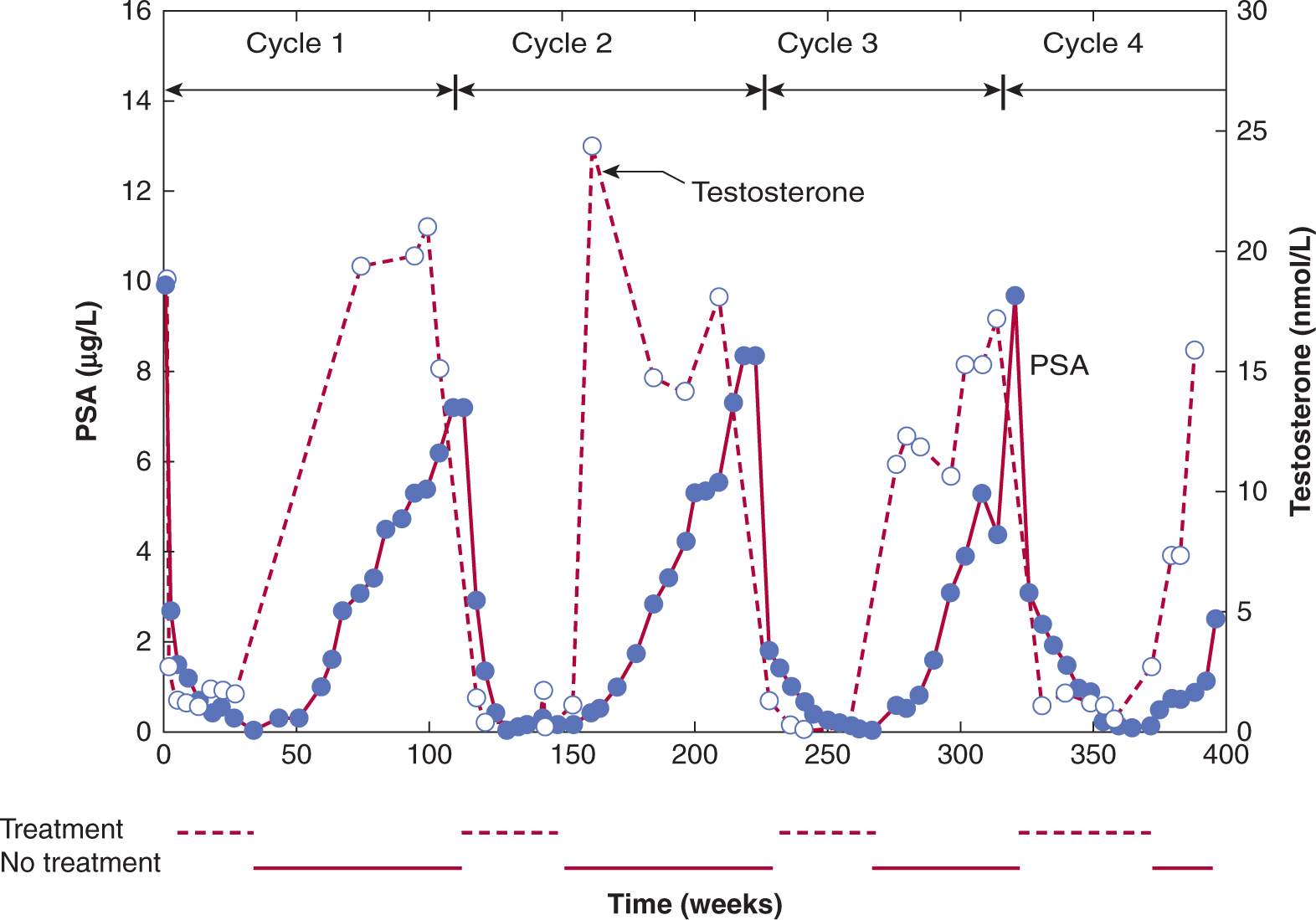
FIGURE 20–14 Intermittent androgen suppression. Approximately 8 years after radical prostatectomy and radiation treatment for positive surgical resection margins, the patient was started on a regimen of intermittent androgen suppression when his serum PSA had increased from a nadir of 0.6 μg/L to 10.4 μg/L. He was treated with a combination of antiandrogen (cyproterone acetate) and LHRH agonist (leuprolide acetate). The patient has undergone 4 cycles of androgen withdrawal and replacement over a period of more than 7 years. Open circles indicate serum testosterone values (nmol/L) and closed circles are serum levels of PSA (μg/L). (Used with permission from Dr. N. Bruchovsky.)
The effectiveness of intermittent androgen suppression and replacement for delaying progression to androgen independence was first shown using the androgen-dependent Shionogi mouse mammary carcinoma (Akakura et al, 1993). With this tumor model, complete remissions are observed after androgen withdrawal, but invariably the disease recurs and is refractory to further hormonal manipulations. Because progression to androgen independence was linked to the cessation of androgen-induced differentiation of these stem cells, the effect of replacement of androgens at the end of a period of regression was tested by transplantation into noncastrated males (Akakura et al, 1993). This cycle of transplantation and castration-induced apoptosis was repeated and relative to onetime castration, intermittent androgen suppression approximately tripled the time to androgen independence. Other experimental models of hormone-dependent prostate cancer have confirmed these effects.
Prospective clinical trials during the past 10 years have evaluated intermittent hormonal therapy and have found that intermittent treatment is at least as effective as continuous androgen suppression, while showing reduced treatment-related toxicity, lower cost, and improved quality of life, including recovery of sexual potency.
Androgen-deprivation therapy has been investigated before or after primary treatments for localized prostate cancer, such as prostatectomy or radiation therapy, or concurrent with radiation therapy. Initial hormone treatment can lead to a reduction in tumor size, but such therapy has not been shown to improve survival following radical surgery. However, the combination of radiation and hormonal therapy for locally advanced prostate cancer does prolong survival (Bolla et al, 2002; Warde et al, 2011).
20.4.6 New Hormone-Biological Therapies for Prostate Cancer
As described in Section 20.5.5, there may be a role for targeting those genes critical to cell survival (eg, TRPM-2, BCL-2, IGFBP-5) as enhancement of apoptotic cell kill might enhance the effects of androgen withdrawal and thereby delay time to recurrence and emergence of androgen independence. Similarly, expression of the cell-survival protein clusterin is increased in prostate cancer in response to many treatments, including hormone ablation therapy, chemotherapy, and radiation therapy. Increased clusterin production is linked to faster rates of cancer progression, treatment resistance, shorter survival duration, and is closely correlated with increasing grade (ie, less differentiation), which is a strong prognostic factor for poor survival of patients with prostate cancer; thus clusterin is a possible target for tumor therapy. Several preclinical studies using androgen-regulated tumor models have demonstrated proof of principle that administration of antisense oligodeoxy nucleotides is a viable approach to reducing activity of this and other survival factors, and these agents are being evaluated in clinical trials.
Understanding that progression to CRPC often occurs despite continued dependence on signaling from the AR (see Sec. 20.5) opens several doors to new therapeutic approaches, such as steroid enzyme inhibitors and more potent antiandrogens. Each enzyme involved in the androgen synthesis pathway, such as CYP17A1, may be a target for drug discovery. These agents could then be used in combination with existing AR antagonists and might increase the survival of patients.
20.4.7 Hormone and Chemoprevention Strategies for Prostate Cancer
Epidemiological studies of prostate cancer suggest that diet and lifestyle probably contribute more to prostate carcino-genesis than racial or familial factors. The observation that premalignant prostatic lesions (PIN) occur with almost equal frequency in different racial populations with both high and low risk of prostate cancer implies that a limiting step is progression from subclinical to locally invasive carcinoma. Also, there is considerable evidence that androgen stimulation over a long period of time is a necessary prerequisite for prostate cancer and therefore an obvious target in any chemoprevention strategy.
Clinical trials using tamoxifen as a chemopreventive agent for breast cancer in high-risk groups of women have provided a paradigm for using drugs that block androgen action and thereby prevent the emergence of prostate cancer. Drugs that show promise are the antiandrogen bicalutamide and the 5α-reductase inhibitors finasteride and its analog dutasteride (see Fig. 20–13), which block the intracellular metabolism of testosterone to the more potent DHT (see Fig. 20–3). The toxicity of antiandrogens such as bicalutamide (gynecomastia, gastrointestinal toxicity, etc) poses concerns for application in prevention studies, but the toxicity profiles of finasteride and dutasteride are more favorable. A Phase III trial of finasteride in 18,000 men older than 50 years of age has shown a cumulative reduction of 25% at 7 years of treatment in early stage, organ-confined, low-grade prostate cancer, but there was concern that use of finasteride was associated with increased prevalence of higher-grade disease (Thompson et al, 2003). A retrospective study of men involved in this study showed that finasteride may have contributed a bias toward the detection of high-grade disease by improving the performance characteristics of PSA and of prostate biopsies (Elliott et al, 2009). Similarly, a 4-year study showed that men receiving a daily dose of dutasteride reduced their incidence of developing cancer (although it did not reduce the development of the more lethal high-grade cancers) and improved the outcomes related to BPH (Andriole et al, 2010).
20.5 MECHANISMS FOR PROGRESSION TO HORMONE INDEPENDENCE AND DEVELOPMENT OF RESISTANCE TO HORMONAL THERAPIES
Cancer progression involves a series of changes in the malignant cell whereby its appearance and behavior inevitably evolve toward a more aggressive and poorly differentiated phenotype (see Fig. 20–8; see Chap. 10, Sec. 10.1). In breast and prostate cancer, the term progression is more commonly used in an endocrine sense to connote the process by which there is a change from hormone-dependent to the hormone-independent phenotype.
Resistance to hormonal therapies is of 2 types: de novo/intrinsic and acquired. Mechanisms responsible for de novo and acquired resistance to tamoxifen may differ because ER+ breast tumors with de novo tamoxifen resistance also are generally resistant to other forms of endocrine therapy. In contrast, tumors with acquired tamoxifen resistance are more likely to respond subsequently to second- and third-line endocrine therapy. This observation suggests that although some mechanisms of endocrine therapy resistance are general, there will be mechanisms specific for individual therapies (Musgrove and Sutherland, 2009). Less is known about resistance to aromatase inhibitors, but similar to tamoxifen resistance, multiple mechanisms are likely to be involved (Miller, 2010). Potential mechanisms include altered pharmacokinetics and pharmacogenetics of aromatase, decreased ER expression and/or activity together with increased activation of alternate signaling and survival pathways such as insulin-like growth factor receptor (IGFR) or EGFR (Johnston et al, 2009; Sabnis and Brodie, 2010). Interestingly, there seem to be some mechanisms common to aromatase inhibitors generally and others unique to individual aromatase inhibitors as crossresistance is not always seen and there are few clinical correlative data available to determine the relevance and/or frequency of such mechanisms in vivo (Miller, 2010). Figure 20–15A and B summarize potential mechanisms thought to be involved in resistance to tamoxifen and aromatase therapy, respectively.
FIGURE 20–15 Summary of the multiple mechanisms of resistance to (A) tamoxifen and (B) aromatase inhibitors in human breast cancer. REA, repressor of ER activity; PTM, post-translational modifications
20.5.1 Clonal Selection Versus Adaptation
Tumor progression is attributed usually to mutations, or to irreversible chromosomal rearrangements, losses, and duplications, with selection of clones that have a growth or survival advantage. This widely accepted mechanism proposes that androgen or estrogen-independent tumor cells are present initially and that hormonal therapy kills all but this population of cells, which subsequently becomes the dominant phenotype of the tumor. There is evidence to support this mechanism in some animal tumor models whose rapid regrowth after androgen withdrawal implies the preexistence of androgen-independent clones (Gingrich et al, 1997). However, there is evidence to support alternative mechanisms, including heritable perturbations in regulatory pathways, that depend on interactions with inter- or intracellular factors, and which may be in part a result of epigenetic/adaptive processes. Figure 20–16 outlines the 2 mechanisms for progression.
FIGURE 20–16 Clonal selection and epigenetic/adaptive mechanisms for emergence of hormone independence in tumors. A) Upon hormone withdrawal, hormone-dependent cells are killed, leaving behind preexisting hormone-independent clones that repopulate the tumor. B) Upon hormone withdrawal, most cells are killed except for those expressing critical cell-survival genes. When reexposed to hormone, the pattern of gene expression reverts and the tumor regrows in response to hormone. However, through further adaptive changes in gene expression patterns, the cells no longer require hormone to sustain their growth.
For breast cancer there are molecular data showing identical genetic abnormalities in synchronous premalignant, preinvasive, and invasive breast cancer lesions that support clonal selection in breast cancer progression. However, unlike prostate cancer where the vast majority of invasive prostate cancers are AR+, 2 biologically distinct groups of breast cancers exist, that is, ER+ and ER– tumors. Infrequent changes in ER status during tumor progression, suggest that the 2 tumor types are unlikely to be derived one from the other. Furthermore, when ER status has changed during progression, it has been impossible to distinguish between the suppression of ER expression due to promoter methylation, or that the original primary tumor was heterogeneous containing both ER+ and ER– breast cancer cells, with ER– cancer cells having a growth advantage. Most evidence suggests that progression to hormone independence and development of tamoxifen and/or aromatase inhibitor resistance in breast cancer is a result of epigenetic and adaptive mechanisms. However, discordance of receptor status between primary and metastatic breast cancer is frequent enough and has sufficient clinical implications to recommend biopsy and assessment of the receptor status of metastatic tumors where possible (Amir et al, 2012).
There are many molecular processes linked to prostate cancer progression that imply an epigenetic mechanism (Rennie and Nelson, 1998). Early experiments demonstrated that chronic administration of combinations of testosterone and estradiol to niobium (Nb) rats increased the incidence of prostate adenocarcinomas from less than 1% to greater than 18%. As neither estrogens nor androgens are mutagens, this finding suggests that prostate cancer can arise through an epigenetic mechanism. Furthermore, in this animal model, fractional replacement of estrogen by tamoxifen to keep the tumor in stasis (ie, neither regressing nor growing) delayed or prevented progression to hormone independence, implying that progression was also driven by epigenetic processes.
The extent to which the development or progression of breast and prostate cancer are driven by epigenetic or genetic mechanisms has important clinical implications. If progression occurs primarily as a consequence of preexisting genetic changes, perhaps in putative stem cells (see Chap. 13, Sec. 13.4), then options for prevention and treatment are limited. Conversely, if reversible adaptive or epigenetic processes are predominant, then breast and prostate cancer are potentially more amenable to therapeutic control, as indicated in Figure 20–16.
20.5.2 Altered Pharmacology
A possible mechanism for tamoxifen resistance in breast cancer is altered uptake and metabolism of tamoxifen by the tumor leading to reduced intratumoral drug concentrations. Because CYP2D6 is the major enzyme responsible for the generation of endoxifen, the active metabolite of tamoxifen (see Fig. 20–11), individuals with CYP2D6 gene polymorphisms that reduce enzymatic activity may have reduced benefit from tamoxifen (Hoskins et al, 2009; Kiyotani et al, 2010), although not all studies agree with this conclusion. Differences in the frequencies of mutations in the CYP2D6 gene occur in different ethnic groups and may influence the effectiveness of tamoxifen (Bernard et al, 2006). Also, selective serotonin reuptake inhibitors (SSRIs), and/or selective noradrenaline reuptake inhibitors (SNRIs) are prescribed quite commonly for depression or other reasons and these drugs are known to inhibit CYP2D6 activity (Hoskins et al, 2009). Altered availability of tamoxifen to ER within the tumor may also occur as a result of increased expression of binding proteins such as antiestrogen binding sites (AEBSs) that do not bind estrogen but would sequester tamoxifen away from ER. Data consistent with these mechanisms have been described both in some human breast tumors and in a model of tamoxifen-resistant human breast cancer where breast cancer cell lines are grown as xenografts in athymic mice (Clarke et al, 2001).
20.5.3 Alternative Signal Transduction Pathways and the Importance of Stroma
During progression to hormone independence, both autocrine and paracrine growth factor pathways may become dysregulated and growth factors may replace estrogens or androgens as primary growth regulators. Both normal and neoplastic epithelial cells exist in a complex 3D interaction with stromal cells (fibroblasts and adipocytes), blood vessels, and often immune cells within and surrounded by an extracellular matrix (ECM). Cell–cell interactions may be mediated by growth factors or through interactions with the ECM via adhesion molecules called integrins located on the cell surface (see Chap. 10, Sec. 10.2). These interactions regulate signal transduction pathways and gene expression.
20.5.3.1 Alternate Pathways of Signal Transduction in Breast Cancer In breast cancer, ligand-independent activation of the ER has been demonstrated frequently, and may contribute to hormone independence and antiestrogen resistance. Some of the more common alterations in breast tumors are upregulated and abnormally regulated growth factor pathways (EGFR, IGFR, HER2/NEU/ERBB-2) and/or their intracellular signal transduction molecules (RAS/RAF, MAPK, phophatidylinositol-3 kinase [PI3K], AKT; see Chap. 8, Sec. 8.2). Growth factors such as IGF-I, heregulin and EGF can activate ER in the absence or presence of estrogen through mechanisms involving phosphorylation of coregulators or of the ER itself (Musgrove and Sutherland, 2009). Similar phosphorylation mechanisms are almost certainly operational for the ligand-independent activation of ER by protein kinase A, protein kinase C, pp90rsk1, and protein kinase B (Ali and Coombes, 2000; Clarke et al, 2001). The expression and/or activity of many of these kinases are often increased in breast tumors compared to normal breast tissue, and specific phosphatases that deactivate these kinases are expressed at higher levels in some breast cancer cell lines with altered responses to estrogen. Thus an altered phosphorylation profile of the ER and/or its coregulators may underlie the progression to hormone independence and endocrine resistance (Xu et al, 2009; Skliris et al, 2010). The ability of tamoxifen to effect ligand-independent activation of ER is variable, which suggests that this is only one of several mechanisms that participate in the development of antiestrogen resistance (Clarke et al, 2001). For example, sustained activation of the PI3K/AKT pathway (see Chap. 8, Sec. 8.2.5) can protect breast cancer cells against tamoxifen-induced apoptosis. This is because of a positive feedback loop, whereby activated AKT activates ERα in a ligand-independent fashion, and ERα in the presence or absence of estrogen activates PI3K (Campbell et al, 2001; Sun et al, 2001). Because growth factors activate both the PI3K and MAPK pathways and both can impact ERα signaling, it seems that a marked amplification of ligand-independent ER signaling is inevitable.
Crosstalk between ER and other transcription factors or coregulator proteins may also play a role in progression to hormone independence. For example, crosstalk between ER and the transcription factor AP-1 takes several forms: estrogens can regulate the expression of the components of AP-1 transcription complexes; ER and AP-1 can interact directly by protein–protein interactions to regulate target gene expression; activation of AP-1 complexes can downregulate ER expression (Clarke et al, 2001). A mouse model of tamoxifen-resistant breast cancer has also shown an association of resistance with oxidative stress and increased AP-1 activity (Schiff et al, 2000). All of these mechanisms could influence the responsiveness of a breast cancer cell to tamoxifen as well as to aromatase inhibitors, and there are some reports where altered AP-1 activity and/or levels of expression of AP-1 components can be correlated with acquired tamoxifen resistance. Thus multiple mechanisms may lead to progression to hormone independence and the development of resistance to endocrine therapies (see Fig. 20–15). Increasing knowledge of such mechanisms is leading to clinical trials evaluating endocrine therapy in combination with other therapies targeted to key molecules within signaling pathways known to be active within the patient’s tumor (Di Cosimo and Baselga, 2010).
20.5.3.2 Alternate Pathways of Signal Transduction in Prostate Cancer In prostate cancer, there is an increase in paracrine stimulation by growth factors produced in prostatic stroma with eventual autocrine production and stimulation by the prostate cancer epithelial cell (Rennie and Nelson, 1998). For example, in BPH, epithelial cells express EGFR but not its ligand TGF-α, whereas prostate stroma produces TGF-α but not EGFR. However, in many prostate epithelial tumor cells, coexpression of EGFR and TGF-α is observed, indicating a shift from paracrine to autocrine stimulation (Leav et al, 1998). Whether the molecular mechanisms responsible for the shift in this regulatory loop are a result of mutational or adaptive processes is unknown. However, by simply adapting the amount of the tyrosine kinase inhibitor genistein in the diet, the expression of EGFR in the EGFR/TGF-α system could be manipulated in a rat prostate cancer model (Dalu et al, 1998).
A similar shift from paracrine to autocrine regulation has been seen with the IGF regulatory network. In the normal prostate and in BPH, IGF-I, and IGF-II are produced and secreted by the stroma, and influence prostate epithelia in a paracrine fashion through IGF-I and IGF-II receptors. However, in prostate cancers, production of both types of IGF occurs in the tumor cells, which results in an autocrine loop for androgen-independent growth stimulation (Wang and Wong, 1998). Overall, as prostate cancer progresses, the stromal component tends to become redundant for controlling prostate growth.
There is evidence that intratumoral androgen synthesis occurs in men after chemical or surgical castration for prostate cancer (Fig. 20–17). Expression of genes mediating androgen synthesis in the prostate (eg, AKR1C2 and AKR1C1 involved, respectively, in the production of 3α-androstanediol and 3β-androstanediol from 5α-DHT), is elevated or modified in CRPC in contrast to primary tumors (Knudsen and Penning, 2010). In addition, testosterone levels in CRPC cells have been shown to be adequate for activation of androgen-dependent genes. Abiraterone acetate, an inhibitor of the CYP17A1 hydrolase involved in the transformation of progesterone to androstenedione, reduces the plasma level of testosterone in CRPC patients whose levels of circulating testosterone are already in the castrate range (Attard et al, 2008), and probably also inhibits testosterone synthesis in prostatic tissue. As indicated above, this drug leads to tumor response in a substantial proportion of men with metastatic CRPC, and improves their survival, indicating that nonlocalized tumors are often still dependent on androgens (Knudsen and Penning, 2010).
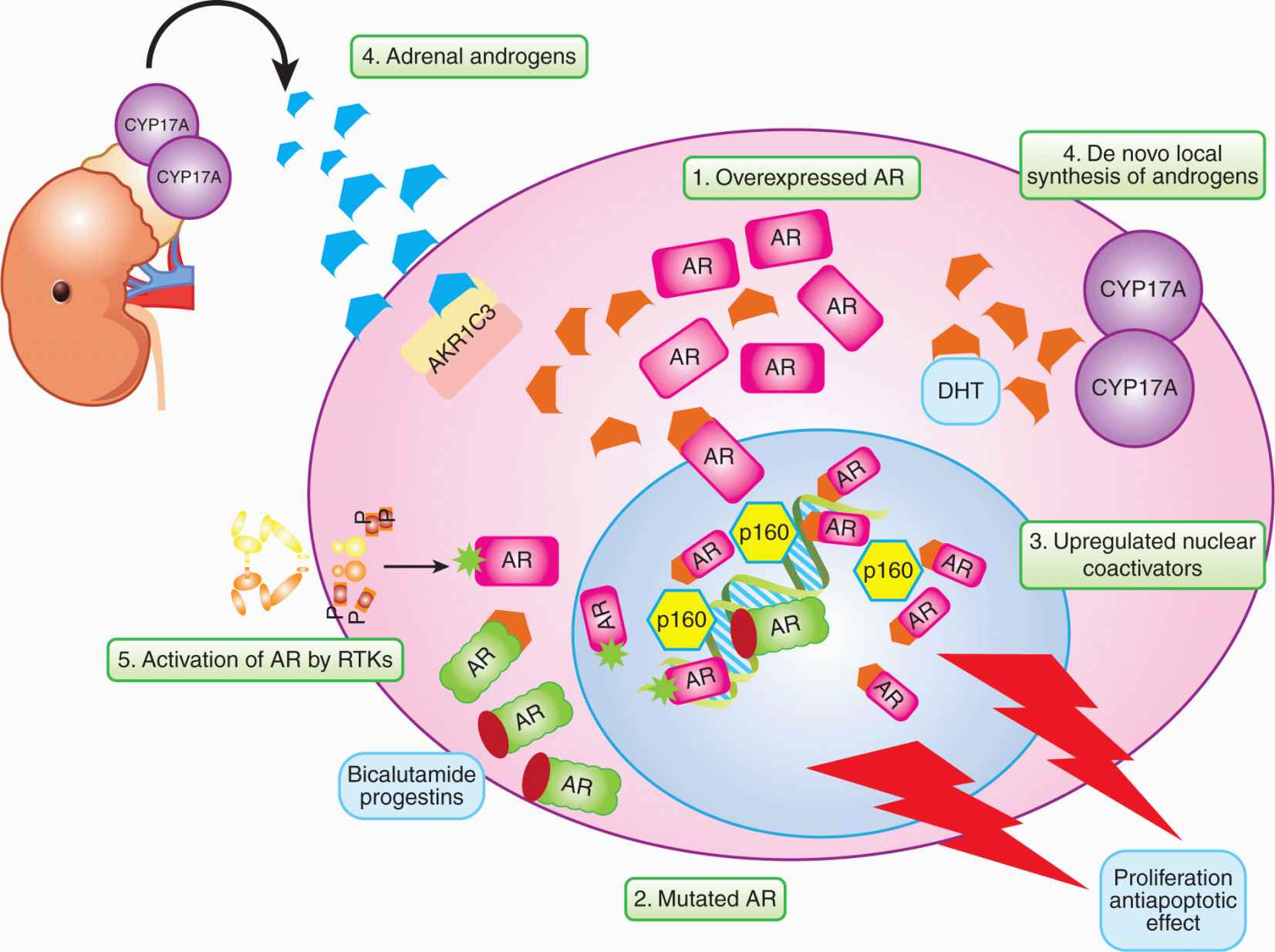
FIGURE 20–17 Summary of the multiple mechanisms of castration-resistant prostate cancer. Key regulatory proteins are the androgen receptor (AR), cytochrome P450 17A1 (CYP17A), p160 family members including SRC-1 and TIF2/SRC-2 (P160), aldo-keto reductase (AKR) 1C family member 3 (AKR1C3), and Receptor Tyrosine Kinase (RTK).
Many molecular alterations in prostate cancer have been discovered in recent years. Foremost among many are gene fusions involving E-twenty-six (ETS) genes, and especially the estrogen-regulated gene, ERG, and the androgen-regulated, prostate-specific, transmembrane protease serine 2 (TMPRSS2) gene, which can be found in approximately 50% of prostate cancers. In addition, several other ETS gene rearrangements have been discovered, including rearrangement of ETV1, ETV4, and ETV5 with TMPRSS2, albeit at much lower frequencies than seen for ERG. TMPRSS2 had previously been characterized as an androgen-regulated gene and thus its androgen-responsive regulatory element drives ERG overexpression in fusion-positive cases, promoting proliferation, invasion, and motility. It has been shown that androgen-dependent signaling induces proximity of the TMPRSS2 and ERG genomic loci, facilitating the formation of the TMPRSS2-ERG gene fusion in response to genotoxic stress (Mani et al, 2009). Because prostate cancer is mainly driven by androgen signaling, this may explain why these types of fusions are apparently restricted to prostate cancer. Additional ETS gene rearrangements are being discovered, such as between the androgen-inducible tumor-suppressor NDRG1 (N-MYC downstream regulated gene 1) and ERG, while TMPRSS2 rearrangements that do not involve known ETS genes have also been identified, suggesting that other androgen-response oncogenic elements may yet be discovered. These ETS fusions are being studied as biomarkers for prostate cancer initiation and/or progression, but correlation of their presence with clinical outcome remains controversial.
The critical genetic events associated with progression of breast and prostate cancer are still poorly understood, but owing to the development of approaches like gene microar-rays, candidate genes are being discovered at a rapid rate. The current view is that the more likely mechanisms for progression of breast and prostate cancer involve ligand-independent activation of AR or ER via crosstalk with other transcription factors, signal transducers, and related pathways.
20.5.4 Estrogen Receptor and Androgen Receptor Amplifications and Mutations
In breast cancer, de novo hormone independence is often associated with absence of ER. However, a substantial proportion of ER+ breast tumors are either resistant de novo to endocrine therapy or acquire resistance after treatment with endocrine therapies despite the continued expression of ER. The presence of ER mutations and/or amplifications in breast cancer is infrequent. Although mutated ERα is quite rare in human breast cancers, variant ERs generated by alternative RNA splicing of both ERα and ERβ genes are common. Changes in expression of some of these variants have been documented in breast tumors, but only limited correlation with endocrine resistance has been reported (Shi et al, 2009).
Unlike breast cancer, androgen-independent human prostate cancer is seldom associated with absence of AR. Immunohistochemical analysis of AR in biopsies from virtually every stage and grade of prostate cancer show that AR is retained regardless of hormone sensitivity. Only in some multiply-passaged androgen-independent human prostate cancer cell lines, such as Du145 and PC3 cells, is there lack of detectable or functional AR. Because loss of AR expression is not normally associated with the malignant phenotype, attention has focused on more subtle mutational events in the AR gene that could give rise to alterations in AR activity (see Fig. 20–17).
Estimates of the incidence of somatic point mutations in the AR gene range from 0% to 30% of patients with CRPC and there is a tendency for the incidence of AR mutations to occur more frequently in advanced or metastatic disease. Also, patients treated with antiandrogens are more likely to have AR mutations as compared to patients treated solely by surgical castration or LHRH agonists (Taplin et al, 1999). Many AR mutations in the ligand-binding domain have been characterized, showing a range of effects from partial or complete loss of function as well as diverse transactivational activity (Shi et al, 2002). The well-characterized threonine-to-alanine (T877A) mutation in the AR ligand-binding domain of the LNCaP human prostate cancer cell line has been shown to alter androgen binding specificity, such that it can be activated by high concentrations of virtually any steroid or antiandrogen (Duff and McEwan, 2005). Similar types of mutations have been observed in some patients who manifest the anti-androgen withdrawal syndrome. Truncated forms of the AR expressed as alternatively spliced variants lacking a ligand-binding site are also constitutively active in some men with CRPC (Hu et al, 2009).
Amplification and overexpression of the wild-type AR gene are likely more common than mutations. Studies before and after androgen ablation have shown gene amplification of wild-type AR in approximately 30% of recurrent tumors. In AR gene-amplified tumors, PSA immunostaining appears to be about twice as high as in tumors with no amplification, indicating that AR gene amplification leads to upregulation of the PSA gene (and possibly other androgen-regulated genes). Thus patients with AR gene amplification may have elevated serum PSA concentrations without a clear correlation with tumor burden (Koivisto and Helin, 1999). The AR is the only gene consistently upregulated in tumor progression (Chen et al, 2004), and although androgen withdrawal is ineffective in most castration-resistant prostate tumors, knockdown of AR using small interfering RNAs was able to decrease serum PSA and, in some cases, to cause tumor regression (Snoek et al, 2009).
20.5.5 Altered Receptor Structure and Function, Including Altered Coregulator Activity
The ability to recruit coactivators and/or corepressors to a promoter plays an important role in transcriptional regulation by a steroid hormone (see Fig. 20–4). Experimental alteration of the relative expression and/or activity of coactivator to core-pressor suggests that such a mechanism can influence how a target cell interprets tamoxifen, either as an antiestrogen or an estrogen. Alterations of specific coactivators and corepressors of ER have been described in human breast tumors and correlations with resistance to tamoxifen have been reported (Osborne et al, 2003; Musgrove and Sutherland, 2009). Because coregulators are essential to ER signaling, altered expression and/or activity could theoretically affect sensitivity of tumors to aromatase inhibitors as a result of estrogen independence of growth and survival pathways. Growth factor/phosphorylation pathways can also influence steroid hormone receptor pathways via their ability to modulate coactivators by phosphorylation (Han et al, 2009) and may influence on the emergence of hormone independence.
In prostate cancer, overexpression of coactivators might lower the ligand threshold of the AR such that physiological concentrations of adrenal androgens are then adequate to drive androgen-regulated responses, such as PSA expression (Chmelar et al, 2007). Indeed, immunohistochemical evaluation of human prostate cancer samples demonstrated overexpression of the coactivator SRC-1 in approximately half of nontreated prostate cancers, as well as high levels of both SRC-1 and SRC-2 in the majority of castration-resistant tumors (Gregory et al, 2001). Other coactivators that are involved in prostate cancer progression and in the development of a castration-resistant state are the thyroid hormone receptor-associated protein mediator complex (MEDI/TRAP220), which is involved in AR-mediated transcription of the PSA gene, and AR chaperones such as heat shock protein (HSP) 27 and cell division cycle (CDC) 37. Chaperones have an essential role in stabilizing AR interaction with DHT. Interestingly, MED1/TRAP220 is overexpressed in both AR-positive and -negative prostate cancer cell lines, as well as in clinically localized prostate cancers, suggesting that MED1/TRAP220 hyperactivity might promote prostate tumor formation.
20.5.6 Changes in Gene Expression
The inappropriate upregulation of cell-survival genes is another potential mechanism whereby cancer cells become treatment-resistant. Recently, genome-wide analyses and the generation of gene expression signatures have added support to experimental data that deregulation of proliferation and survival pathways plays an important role in resistance to both tamoxifen and aromatase inhibitors (Musgrove and Sutherland, 2009).
The BCL family of genes has been implicated in progression of breast cancer and BCL-2 is an estrogen-inducible gene in ER+ breast cancer cell lines. Other survival pathways, such as the AKT (protein kinase B) pathway, may also be important in progression of human breast cancer. As described above, AKT is activated by ligand-bound EGFR and IGFR, and can phosphorylate coactivators as well as ERα (Musgrove and Sutherland, 2009), and can activate ER in a ligand-independent fashion. Furthermore, overexpression of AKT in breast cancer cell lines leads to upregulation of BCL-2 and protects breast cancer cells from tamoxifen-induced apoptosis (Campbell et al, 2001). There appears to be a positive feedback loop involved in this network whereby ERα in the presence or absence of estrogen can activate the regulatory subunit of PI3K, which, in turn, activates AKT (see also Sec. 20.5.3.1).
Tumor-suppressor genes, such as Rb (retinoblastoma) and p53, are inactivated in many tumor types, including breast and the late stages of prostate cancer. E-cadherin and other cell-adhesion genes, which have been characterized as suppressors of the metastatic phenotype, are frequently inactivated or downregulated during progression to advanced prostate cancer and are associated with poor clinical outcome (Bussemakers, 1999).
In prostate cancer, elevated expression of the antiapoptotic gene BCL-2 is found in virtually all CRPC and overexpression confers resistance to androgen withdrawal by blocking the normal apoptotic signals (McDonnell et al, 1997). The anti-apoptotic protein clusterin (also known as TRPM-2 or SGP-2) is also upregulated with disease progression in prostate cancer; it confers resistance to hormonal therapy when overexpressed in prostate tumor cell lines (Steinberg et al, 1997).
Related posts:
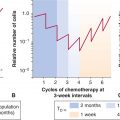
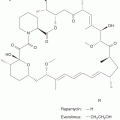
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


