HORMONAL CONTROL OF CALCIUM HOMEOSTASIS
A complex homeostatic system has evolved that is designed to maintain near constancy of Ca2+o through calciotropic hormone, and direct, Ca2+o-induced alterations in the GI, renal, and/or skeletal handling of Ca2+ (Fig. 49-2).4,5,6 and 7 Usually, this is accomplished in such a way that extracellular (including skeletal) and cellular Ca2+ homeostasis and balance are maintained simultaneously. Conditions of gross Ca2+ excess, however, may induce positive Ca2+ balance (e.g., deposition of Ca2+ in bone and soft tissues) to maintain a normal or near-normal level of Ca2+o.Conversely, severe Ca2+ deficiency leads to mobilization of skeletal Ca2+ to preserve normocalcemia. Overall Ca2+o homeostasismay be understood in terms of the following general principles: (a) The first priority of the homeostatic system is to maintain a normal level of Ca2+o; (b) with moderate stresses on the system, intestinal and renal adaptations are usually sufficient to sus- tain Ca2+o homeostasis without changes in net bone mass; and (c) with severe hypocalcemic stresses, skeletal stores of Ca2+ are mobilized to maintain Ca2+o homeostasis, potentially compromising the structural integrity of the skeleton.
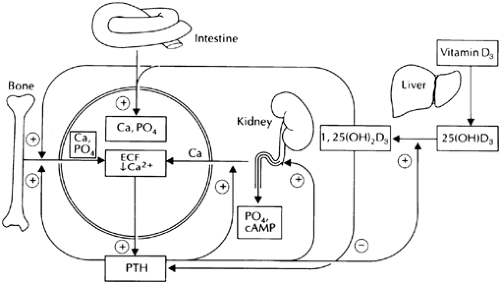 FIGURE 49-2. The hormonal control of calcium and phosphate homeostasis by parathyroid hormone (PTH) and vitamin D (solid lines and arrows). Ca2+ regulates PTH secretion in an inverse fashion. In turn, PTH affects renal handling of calcium and phosphate as well as renal synthesis of 1,25(OH)2D3. PTH and 1,25(OH)2D3 synergistically mobilize calcium and phosphate from bone, whereas the latter increases intestinal absorption of both ions. Also shown are direct actions of calcium and phosphate ions on tissues involved in maintaining mineral ion homeostasis (lines and arrows), illustrating the role of these ions as extracellular, first messengers. For example, elevated Ca2+o directly suppresses renal synthesis of 1,25(OH)2D3, tubular reabsorption of Ca2+ in the thick ascending limb, and the resorptive activities of osteoclasts. Thus, calcium ions modulate their own homeostasis not only through direct actions on the secretion of calciotropic hormones but also by exerting direct effects on target tissues that tend to lower Ca2+o. In effect, calcium and phosphate ions act as Ca2+o-regulating factors or “hormones” because they transmit information about the state of mineral ion metabolism from one part of the homeostatic system to other parts. For additional details, see text. (Reproduced from Brown EM, Pollak M, Hebert SC. Cloning and characterization of extracellular Ca2+-sensing receptors from parathyroid and kidney. Molecular physiology and pathophysiology of Ca2+-sensing. The Endocrinologist 1994; 4:419.) |
The homeostatic system comprises two essential components. The first is several cell types that sense changes in Ca2+o and respond with appropriate alterations in their output of Ca2+o-regulating hormones (PTH, calcitonin, and 1,25[OH]2D3). The parathyroid glands are key sensors of variations in Ca2+o, responding withalterations in PTH secretion that are inversely related to ambient levels of Ca2+o.7 Parathyroid cells recognize changes in Ca2+o through a cell-surface Ca2+o-sensing receptor14—described in more detail later—that is a member of the superfamily of G protein– coupled receptors that includes the receptors for PTH and calcitonin. The C cells of the thyroid gland also respond to changes in Ca2+o with alterations in calcitonin secretion—with high Ca2+o stimulating calcitonin secretion—through the same Ca2+o-sensing receptor. The physiologic relevance of changes in circulating levels of calcitonin in adult humans, however, remains uncertain (see Chap. 53). Finally, the production of 1,25(OH)2D3 is likewisedirectly regulated by Ca2+o, with hypocalcemia stimulating its synthesis in the renal proximal tubule and hypercalcemia inhibiting it.15 The second key component of the homeostatic system is the effector cells that control the renal, intestinal, and skeletal handling of Ca2+. The translocation of calcium and phosphate ions into or out of the extracellular fluid by these tissues is regulated by PTH, calcitonin, and 1,25(OH)2D3 as well as by mineral ions themselves.
The overall operation of the homeostatic system is as follows (see Fig. 49-2). In response to slight decrements in the Ca2+o, forexample, a prompt increase occurs in the secretory rate for PTH. This hormone has several important effects on the kidney, which include inducing phosphaturia, enhancing distal tubular reabsorption of Ca2+, and increasing generation of 1,25(OH)2D3 from 25-hydroxyvitamin D3, or 25(OH)D3.16 The increased circulating levels of this potent metabolite of vitamin D3 directly stimulate absorption of calcium and phosphate by the intestine through independent transport systems.4,5 and 6,17 PTH and 1,25(OH)2D3 also synergistically enhance the net release of calcium and phosphate from bone. The increased movement of Ca2+ into the extracellular fluid from intestine and bone, coupled with PTH-induced renal retention of this ion, normalize circulating levels of Ca2+o, thereby inhibiting PTH release and closing the negative-feedback loop. In addition, 1,25(OH)2D3, formed in response to the action of PTH on the kidney, directly inhibits the synthesis and secretion of PTH,7 contributing to the negative feedback control of parathyroid function by the homeostatic system. Excess phosphate
mobilized from bone and intestine is excreted in the urine via the phosphaturic action of PTH.
mobilized from bone and intestine is excreted in the urine via the phosphaturic action of PTH.
Both calcium and phosphate ions can exert direct effects on various cells and tissues involved in mineral ion metabolism (see Fig. 49-2). For instance, calcium ions not only reduce PTH secretion but also directly inhibit proximal tubular synthesis of15 stimulate the function of osteoblasts,18 and inhibit 1,25(OH)2D3, the function of osteoclasts.19 Phosphate ions reduce 1α-hydroxylation of vitamin D, stimulate bone formation, inhibit bone resorption,4,5,6 and 7 and stimulate various aspects of parathyroid function,20 as described in more detail in the next section. These actions play important roles in mineral ion homeostasis by enabling both the hormone-secreting and effector elements of the homeostatic system to sense changes in the ambient concentrations of mineral ions and to respond in a physiologically appropriate manner. Indeed, by virtue of their direct actions on cells involved in mineral ion metabolism, calcium and phosphate ions can be viewed as serving a hormone-like role as extracellular first messengers.7 Although the cloning of the Ca2+o-sensing receptorthat mediates direct actions of Ca2+o on parathyroid and renalfunction has clarified substantially how cells sense Ca2+o,21 the mechanism underlying phosphate sensing remains obscure.
REGULATION OF PARATHYROID HORMONE SECRETION BY CALCIUM AND OTHER FACTORS
A steep, inverse sigmoidal relationship exists between PTH secretion and Ca2+o (Fig. 49-3)—hence, the exquisite sensitivity ofthe parathyroid gland to Ca2+o that is critical to maintaining stable normocalcemia.7 This sigmoidal function can be described in terms of maximal and minimal secretory rates, midpoint or set-point, and slope at the midpoint (see Fig. 49-3). The maximal secretory rate provides a measure of the acute secretory reserve of the gland in response to a maximal hypocalcemic stress. Maximal secretory capacity increases with parathyroid cellular hyper-plasia, as in primary or secondary hyperparathyroidism.4,5,6 and 7,21 The secretion of PTH from the parathyroid cell apparently is incapable of being totally suppressed, even at very high levels of Ca2+o(see Fig. 49-3). Importantly, this implies that a sufficient mass of even normal parathyroid tissue could cause hypersecretion of PTH with resultant hypercalcemia. This has, in fact, been documented in rats by transplanting sufficient numbers of normal rat parathyroid glands to cause hypercalcemia in the recipient animals.22 This may, in part, contribute to the hypersecretion of PTH in hypercalcemic hyperparathyroidism The setpoint of normal human parathyroid cells in vitro is ˜1.0 mmol/L of ionized Ca2+and is close to the ambient level of Ca2+o in humans (1.1–1.3mmol/L).7 This parameter is important in determining the level at which the Ca2+o is set by the homeostatic system (the setpointfor Ca2+o is slightly higher than that for the parathyroid cell per se, also being a function of the tissues that are targets for the actions of PTH). In primary hyperparathyroidism, the setpoint of pathologic parathyroid cells is increased to 1.2 to 1.4 mmol/L Ca2+, or sometimes higher, and the serum Ca2+ concentration is correspondingly reset to an elevated level7,21 (see Chap. 58). The steep slope of the relationship between PTH release and Ca2+oensures a large change in secretory rate for a small change in Ca2+o and is important in maintaining the serum Ca2+ concentra- tion within a narrow range. In addition to its sensitivity to Ca2+o,the parathyroid cell alters its secretory rate within seconds to a perturbation in Ca2+o and may also be responsive to other variables, such as the rate and direction of the change.23,24
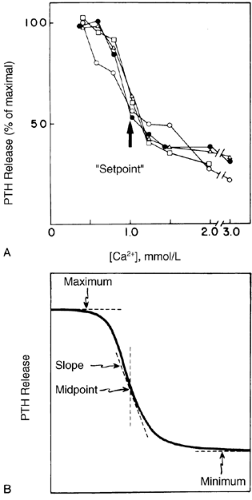 FIGURE 49-3. A, Relationship between parathyroid hormone (PTH) secretion and extracellular Ca2+ concentration in normal human parathyroid cells. Dispersed parathyroid cells were prepared and incubated with the indicated ionized Ca2+ concentrations. The PTH released was determined using a radioimmunoassay that recognizes the intact hormone and the COOH-terminal fragments of the molecule. B, The four parameters that can be used to describe the relationship between PTH secretion and the extracellular ionized Ca2+ concentration: maximal and minimal secretory rates at low and high Ca2+ concentrations, respectively; slope of the curve at its midpoint; and setpoint (the level of Ca2+ producing half of the maximal inhibition of PTH release). (A from Brown EM. Regulation of the synthesis, metabolism and actions of parathyroid hormones. In: Brenner BM, Stein H, eds. Contemporary issues in nephrology, vol 2. Divalent ion homeostasis. New York: Churchill-Livingstone, 1983.) |
The technique of expression cloning in Xenopus laevis oocytes enabled isolation of a phosphoinositide-coupled, Ca2+o-sensing receptor from bovine parathyroid gland through which parathyroid, kidney, and other cells sense Ca2+o.14 This receptor has a large amino-terminal extracellular domain that is involved in binding Ca2+o, followed by seven membrane-spanning segments characteristic of the superfamily of G protein–coupled receptors, and a cytoplasmic carboxy terminus (Fig. 49-4). In addition to recognizing Ca2+o, the receptor responds to other polyvalent cations as well, including Mg2+, trivalent cations, and neomycin.14 The possibility exists, therefore, that the Ca2+o-sensing receptor can function as a Mg2+o-sensing receptor, and that some of the directeffects of Ca2+o as well as the toxic actions of aminoglycosides on the kidney are mediated through it.21 The receptor plays a key role in Ca2+o sensing by the parathyroid and kidney because the human homolog of the receptor harbors inactivating or activating mutations in several human genetic diseases that manifest abnormal Ca2+o sensing.21 In familial hypocalciuric hypercalcemia (FHH),25 individuals with heterozygous inactivating mutations of the Ca2+o-sensing receptor have mild to moderate hypercalcemia with inappropriately normal (e.g., nonsuppressed) circulating levels of PTH and normal or even frankly low levels of urinary calcium excretion. In contrast, persons with homozygous FHH, which presents clinically as neonatal severe hyperparathyroidism (NSHPT), have severe hypercalcemia with marked hyperparathyroidism.25 (NSHPT can also be caused by compound heterozygous inactivating mutations of the Ca2+o-sensing receptor [i.e.,
patients harbor a different mutation in each allele of the receptor26] or by the presence of heterozygous inactivating mutations that exert a dominant negative effect on the wild-type receptor.27) Finally, some families with an autosomal dominant or, occasionally, a sporadic form of hypocalcemia accompanied by relative hypercalciuria (e.g., inappropriately high for the serum calcium concentration) harbor activating mutations in this receptor, a finding that provides further evidence of its importance in Ca2+o sensing in the parathyroid and kidney.28
patients harbor a different mutation in each allele of the receptor26] or by the presence of heterozygous inactivating mutations that exert a dominant negative effect on the wild-type receptor.27) Finally, some families with an autosomal dominant or, occasionally, a sporadic form of hypocalcemia accompanied by relative hypercalciuria (e.g., inappropriately high for the serum calcium concentration) harbor activating mutations in this receptor, a finding that provides further evidence of its importance in Ca2+o sensing in the parathyroid and kidney.28
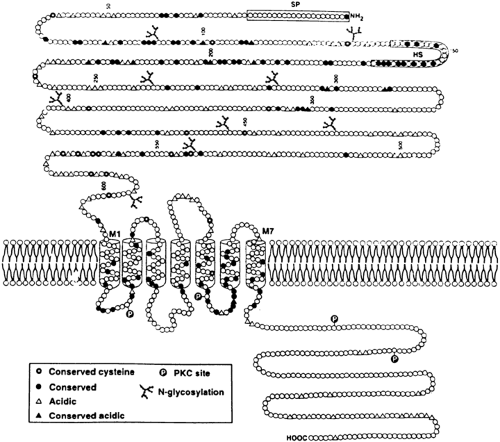 FIGURE 49-4. Proposed structural model of the predicted bovine parathyroid Ca2+-sensing receptor protein. The large amino-terminal domain is located extracellularly and contains nine consensus N-glycosylation sites that are shown as branched chains. Amino acids of the deduced protein are shown at intervals of 50 in the amino-terminal domain. The amino-acid segments that comprise each of the seven predicted membrane-spanning helices are numbered (M1–M7). Potential protein kinase C (PKC) phosphorylation sites are also shown. Each symbol (circle or triangle) represents an individual amino acid; those that are conserved with the metabotropic glutamate receptors are shown as solid symbols, of which acidic residues are indicated by solid triangles and all others by solid circles. Also indicated are multiple cysteine residues conserved with the metabotropic glutamate receptors (which may be involved in stabilizing the large extracellular domain) as well as clusters of acidic residues in the extracellular domain (open and solid triangles) that could be involved in binding of Ca2+ and other polyvalent cations. (SP, signal peptide; HS, hydrophobic segment.) (From Brown EM, Gamba G, Riccardi D, et al. Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature 1993; 366:575.) |
In addition to being present in parathyroid, the Ca2+o-sensing receptor is also present along most of the renal tubule29; it is present at the highest levels in the cortical thick ascending limb of the nephron (a segment that is likely responsible for the abnormal renal handling of Ca2+ in FHH).25 Receptor transcripts are also found in thyroid, brain, and several other tissues, including intestine and bone, as described later.30 The presence of the receptor in kidney probably accounts for several of the long-recognized but poorly understood direct effects of Ca2+o on renal function (such as the reduced urinary concentrating ability encountered in some hypercalcemic patients).31 In the thyroid, the Ca2+o-sensing receptor resides almost exclusively in the C cells and is thought to mediate the stimulatory effect of elevated Ca2+o on calcitonin secretion. Ca2+o-sensing receptors in the brain might mediate as yet unknown actions of Ca2+o on neuronal function.30 Interestingly, among the G protein–coupled receptors, the Ca2+o-sensing receptor exhibits only limited homology with three subclasses of receptors sharing its overall topology, particularly the presence of a very large amino-terminal extracellular domain. These are the metabotropic glutamate receptors (mGluRs), targets for glutamate, the major excitatory neurotransmitter in the brain; the GABAB receptor, which is activated by γ-aminobutyric acid (GABA), the major inhibitory neurotransmitter in the brain; and a family of putative pheromone receptors.30 All three classes of receptor presumably arose from a common ancestral gene. Further understanding of their structural similarities may clarify previously unknown relationships between Ca2+o homeostasis or other functions of Ca2+ in the nervous system and in systemic Ca2+o homeostasis.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


