Hodgkin’s Disease
Hodgkin’s disease is a clonal lymphoid malignancy mainly confined to lymph nodes and lymphoid organs. For the period 1960–1963, 5-year survival from Hodgkin’s disease was 40%; for the period from 1989 to 1993, 5-year survival had increased to 86%.
EPIDEMIOLOGY
About 7500 new cases are diagnosed in the United States each year (roughly 2.9 per 100,000 population) (1). Males are affected somewhat more often than females (M:F 1.4:1). Hodgkin’s disease accounts for about 11% of all lymphomas and is about half as common as multiple myeloma. It has a bimodal age distribution with the first peak in the late twenties and a second peak in late life. The etiology is unknown. Farmers, wood workers, and meat workers are at somewhat increased risk. A minor increased risk is associated with an HLA-linkage disequilibrium. Hodgkin’s disease can complicate the genetic disease and ataxia telangiectasia, and occurs at increased frequency in patients with AIDS. An identical twin of an affected person is at 99-fold increased risk of developing the disease. Some geographic clusters have been noted and molecular studies have implicated Epstein–Barr virus (EBV) in the pathogenesis of some cases, particularly cases in Central and South America and patients with mixed cellularity histology (2) (see below).
PATHOLOGY
Two major forms of Hodgkin’s disease are recognized: classical Hodgkin’s disease accounts for 95% of cases and nodular lymphocyte predominant Hodgkin’s disease accounts for 5% (3). Classical Hodgkin’s disease is divided into four histologic subtypes: nodular sclerosis (70% of cases), mixed cellularity (20% of cases), lymphocyte rich (3%–5% of cases), and lymphocyte depleted (<2% of cases). As diagnostic methods have improved, cases of lymphocyte depleted Hodgkin’s disease have declined both because some cases were actually other lymphoma entities and because earlier diagnosis has made the entity more rare.
The malignant cell of Hodgkin’s disease is the Reed–Sternberg cell; it has different forms in distinct histologic subtypes. In classical Hodgkin’s disease, it is usually derived from a follicular center B cell that has clonally rearranged its immunoglobulin genes but does not transcribe them. Thus, no tumor immunoglobulin molecules are detected. From a clinical perspective, the distinction between classical Hodgkin’s disease and nodular lymphocyte predominant Hodgkin’s disease is critical because the entities differ in natural history and in standard approach to treatment. The distinction between subsets of classical Hodgkin’s disease is not technically difficult, but carries little impact as the natural history and management are not affected by the subset diagnosis.
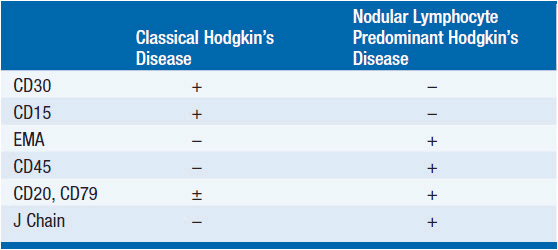
Immunophenotypic studies define differences between classic and nodular lymphocyte predominant Hodgkin’s disease (see Table 32-1). All forms of Hodgkin’s disease share three histologic features: effacement of the normal lymph node architecture; infiltration with a broad range of normal-appearing cells including reactive T cells, plasma cells, histiocytes, neutrophils, eosinophils, and stromal cells (the malignant cells are usually 3% or less of the total cells in an enlarged node); and presence of the characteristic neoplastic cells.
Nodular sclerosis Hodgkin’s disease is the most common form in the United States and is slightly more common in women than men and in the younger age group. The disease grows in nodules separated by bands of collagen fibrosis and the Reed–Sternberg cell is the “lacunar” variant. The name derives from a fixation artifact that causes the cytoplasm to retract leaving a single or multilobed nucleus surrounded by a clear area.
Mixed cellularity Hodgkin’s disease grows in a diffuse pattern and has a florid inflammatory cell background. Reed–Sternberg cells are usually binucleated with prominent nucleoli that give the cell an “owl’s eyes”-like appearance.
Nodular lymphocyte predominant Hodgkin’s disease is characterized by the lymphocyte and histiocyte (L&H) Reed–Sternberg cell variant that is also called a popcorn cell because of a lobulated nuclear contour that resembles popcorn. The cell expresses B-cell markers, surface immunoglobulin, often expresses epithelial membrane antigen (EMA), and cytoplasmic J chains. Unlike the Reed–Sternberg cell in classic Hodgkin’s disease, it is CD30 and CD15 negative. The T cells that cluster around the neoplastic cells often express CD57, a natural killer cell marker and the tumor nodules contain a vague meshwork of CD21+ follicular dendritic cells. It is said that the number of infiltrating CD68+ macrophages influences the risk of relapse; more CD68+ cells correlate with shorter remission duration.
GENETICS
Unlike other lymphoid malignancies, Hodgkin’s disease does not have a characteristic genetic lesion. The cells are aneuploid; Reed–Sternberg cells contain two–eight copies of individual chromosomes (3). A variety of genetic abnormalities have been noted, but none recurs in different cases at high frequency. Cells may contain mutant p53. Evidence for Epstein–Barr virus is noted in 30%–60% of cases, varying with the technique used to detect it. It is commonly present in a clonal episome; however, the only consistent viral gene expressed in the cells containing the EBV genome is LMP1. Its role in the genesis or maintenance of the malignant cells is undefined.
CLINICAL FEATURES OF CLASSICAL HODGKIN’S DISEASE
Patients usually present with painless adenopathy localized to the neck. The mediastinum is involved in the majority of patients (~2/3), occasionally with large masses. When the mediastinal shadow is greater than 1/3, the greatest chest diameter on PA chest radiograph, the mediastinal involvement is considered massive. B symptoms are present in 20%–25%. The disease tends to start in cervical nodes (left side more commonly than right) and march to contiguous lymph node groups. The first intraabdominal site is most often the spleen. Because the spleen has no afferent lymphatics, spleen involvement implies hematogenous spread. The liver is never involved unless the spleen is involved. Involvement of Waldeyer’s ring or epitrochlear nodes is rare. Extranodal involvement is also unusual; when present, bone marrow, liver, lung, pleura, and pericardium are the most commonly involved sites.
In patients with B symptoms, the pattern of the fever may be intermittent. Pel–Ebstein fever describes a pattern in which fever is noted more or less continuously for 1 or 2 weeks followed by afebrile periods of similar duration. However, Pel–Ebstein fevers are unusual. When present, fever tends to occur every evening and breaks while the patient is sleeping giving rise to drenching night sweats.
Pruritis is common, but is not a B symptom. Patients occasionally experience pain in involved lymph nodes upon ingesting alcohol. This is thought to be due to alcohol-induced degranulation of eosinophils. A wide range of symptoms may be noted based on the direct effects of the tumor or paraneoplastic syndromes from tumor products. These symptoms are listed in Table 32-2.

Patients with AIDS who develop Hodgkin’s disease are more likely to have mixed cellularity histology and to have extranodal involvement (2/3 of cases).
CLINICAL FEATURES OF NODULAR LYMPHOCYTE PREDOMINANT HODGKIN’S DISEASE
Patients are predominantly male, in the 30–50-year age group, and usually present with localized peripheral adenopathy involving the cervical, axillary, or inguinal nodes. Mediastinal, spleen, or bone marrow involvement is rare.
DIAGNOSIS AND STAGING
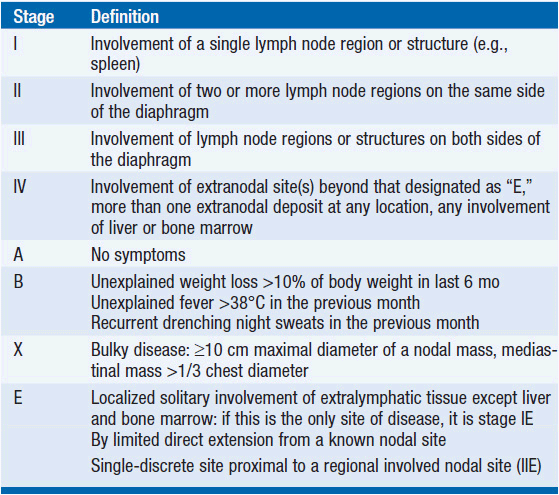
The diagnosis depends on an excisional lymph node biopsy. Needle aspiration is an inadequate diagnostic procedure in a patient with undiagnosed lymphadenopathy. Once a diagnosis is made, a variety of tests are performed to define the extent of disease and the presence or absence of factors that affect prognosis (see Table 32-3). As a result of the testing, the patient is assigned a stage of disease based upon the Cotswolds modification of the Ann Arbor staging classification (see Table 32-4) (4). Because of the orderly progression of Hodgkin’s disease from one lymph node-bearing site to the contiguous node-bearing site, the staging system is an anatomic-based system. However, improvements in treatment over the last 30 years have changed the role of staging in patient management. Stage no longer affects prognosis as primary treatment leads to the cure of about 80% or greater of all patients at all stages of disease. Clinical features other than stage of disease may affect prognosis (Table 32-5).
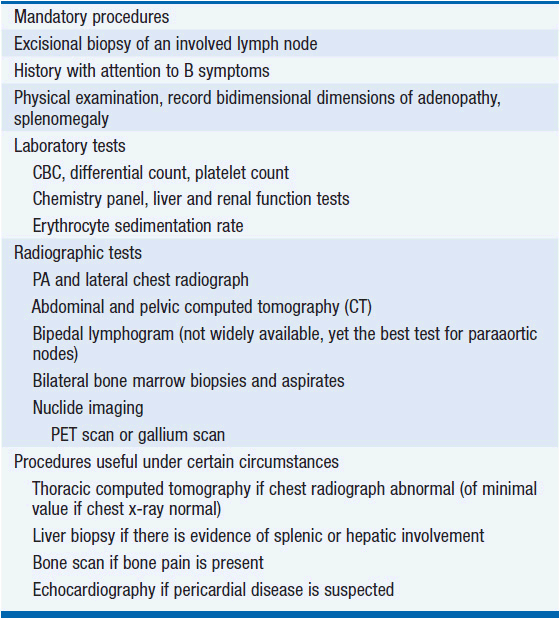

Most patients receive systemic chemotherapy either alone or as part of a combined modality treatment program. Accordingly, the need for precise pathologic staging and exploratory laparotomy has vanished. This change in practice has served to reduce the acute surgical morbidity and mortality risk of the staging laparotomy and to avoid the increased risk of infection associated with splenectomy. Thus, the current state-of-the-art in staging evaluation is to perform clinical staging tests and to include systemic therapy in the management of all patients.
The most poorly evaluated common site of disease is the paraaortic lymph nodes. Bipedal lymphography is more sensitive and specific than abdominal CT in detecting paraaortic node involvement; however, the skill to perform lymphatic channel cannulation is disappearing from radiology departments and, unfortunately, the test is not widely available. However, the widespread use of systemic treatment has resulted in no apparent cost in survival as a consequence of inaccurate abdominal staging.
TREATMENT
 PRIMARY TREATMENT OF NODULAR LYMPHOCYTE PREDOMINANT HODGKIN’S DISEASE
PRIMARY TREATMENT OF NODULAR LYMPHOCYTE PREDOMINANT HODGKIN’S DISEASE
Patients with localized disease are often managed with involved-field radiation therapy (5). The disease may have long periods of remission punctuated by intermittent relapses that do not appear to affect overall survival. The natural history is quite prolonged with survival that is generally as good as or better than classical Hodgkin’s disease. Patients with advanced stage disease are usually managed like patients with classical Hodgkin’s disease (see below). About 3%–5% of patients may undergo histologic progression to diffuse large B-cell lymphoma that is derived from the same malignant clone that gave rise to the Hodgkin’s disease. Such lymphomas are usually responsive to combination chemotherapy and patients are often put into long-term complete remission.
 PRIMARY TREATMENT OF CLASSICAL HODGKIN’S DISEASE
PRIMARY TREATMENT OF CLASSICAL HODGKIN’S DISEASE
Combination chemotherapy is the cornerstone of Hodgkin’s disease treatment. ABVD combination chemotherapy appears to have the best overall efficacy with the least acute and chronic toxicity (6, 7). Clinical staging followed by six cycles of ABVD chemotherapy is certainly a reasonable management approach. Controversy surrounds the use of radiation therapy in the management of Hodgkin’s disease. Many have taken the approach that a shorter course of chemotherapy (e.g., 2 cycles of ABVD) together with 20-Gy mantle-field or involved-field radiation therapy (8) should further improve disease control. While combined modality therapy does somewhat lower the risk of relapse in previously involved lymph nodes, combined modality therapy has not been demonstrated to improve overall survival in early stage (9) or advanced stage disease (10). A key component to assess the impact of treatment is to measure both acute and chronic toxicities.
Radiation therapy to the mediastinum is associated with a threefold increased risk of fatal myocardial infarction (11) and an increased risk of second malignancies in as many as 30% of patients within 30 years of treatment (12). By contrast, randomized comparisons between ABVD versus ABVD plus radiation therapy have not demonstrated significant differences in outcome (13). In the range of doses used to treat Hodgkin’s disease, no convincing evidence has been obtained of a dose-response curve such that lower doses of radiation therapy are less likely to produce late fatal complications. Thus, it is safest to reserve radiation therapy only for the subset of patients who need it to optimize control of their disease.
Who, then, needs radiation therapy? About 5%–12% of patients treated with six cycles of ABVD chemotherapy will not achieve a complete response. This subset of patients benefits from the use of radiation therapy to convert the partial response to a complete response. Patients with low-volume residual disease have a very high rate of conversion to durable complete response. Patients with larger volume residual disease have about a 50% chance of achieving a durable remission. The small group of patients who have a tumor that grows in the face of chemotherapy have a poor prognosis and should be managed with salvage chemotherapy; see below.
Other patients might also benefit from added radiation therapy. Some data suggest that the persistence of PET-positive disease after two or three cycles of chemotherapy may identify patients at risk of relapse if managed with chemotherapy alone. In one series, 80% of patients had no residual PET-positive disease after two cycles of therapy; among the 20%, who had residual PET positivity, two-thirds of those patients relapsed (14). Other data are persuasive that it is PET negativity at the end of treatment that is the superior predictor of outcome (15). Thus, while prospective trials are being performed, it should be possible to define criteria that permit the use of radiation therapy in the subset of patients who have the greatest need for its therapeutic effects while protecting the 80% or so of patients who do well without exposing them to the late toxicities of radiation therapy. Until prospective clinical trial evidence emerges that mid-cycle PET positivity is sufficiently strong a predictor of outcome that one can base treatment decisions on it, the safest approach is to add radiation therapy only to the subset of patients with positive PET scans at the end of their planned course of chemotherapy.
Other regimens have been advocated including Stanford V (16) and BEACOPP (17). Stanford V is a combined modality regimen that involves administering radiation therapy to 100% of patients. It should be kept in mind that it takes 20–30 years to assess the late complications from radiation therapy. Comments about technical advances that should lower the risks are meaningless until a lower risk is actually demonstrated. No one has demonstrated any therapeutic radiation technique that is not associated with late second malignancies. Furthermore, comparison of Stanford V to ABVD in randomized trials suggested that the regimens were comparable in efficacy (18, 19). BEACOPP is a multidrug alkylating agent-based regimen that is said to be superior to COPP/ABVD based on a randomized clinical trial (20). However, BEACOPP is highly myelotoxic, causes infertility and is likely to have an unacceptable late toxicity profile, including secondary leukemia and myelodysplasia, especially because 75% of the patients also received involved-field radiation therapy. Efforts to improve treatment outcome by combining the active agents into 7-, 8-, or 10-drug regimens have not demonstrated clear superiority over ABVD alone.
 SALVAGE TREATMENT
SALVAGE TREATMENT
High-dose chemotherapy with autologous hematopoietic stem-cell transplantation is the cornerstone of salvage therapy for patients with Hodgkin’s disease who relapse or who experience progression during remission induction. The likelihood of success is related to the initial remission duration (21). Those whose initial remission lasted more than 12 months may have a 75%–80% of achieving a durable second remission. Initial remissions shorter than 12 months identify a group of patients that have about a 40%–50% chance of achieving a second durable remission. Patients with progressive disease during induction chemotherapy generally have a 20% or less chance of attaining durable remission.
Salvage treatment is administered in two phases; first a conventional dose regimen to achieve major reduction in tumor bulk and promote mobilization of hematopoietic stem cells into the peripheral blood followed by high-dose therapy with stem cell support. The choice of the conventional dose regimen is usually based on the original remission duration.
In general, patients who experience long initial remissions remain sensitive to the drugs that induced the first remission. However, alkylating agent-based regimens like MOPP (22) or ChlVPP (23) may offer some advantage over a second course of ABVD in the setting of resistant disease (7) as the mechanisms of resistance to natural products like vinblastine and doxorubicin do not appear to influence alkylating agent-based killing. Patients with initial remissions lasting less than 1 year should receive an alkylating agent-based conventional-dose salvage regimen. Patients who experienced progressive disease on ABVD may respond to MOPP or MOPP-like regimens. However, a newer regimen with novel agents such as ESHAP (etoposide, methylprednisolone, high-dose cytarabine, cisplatin) (24) may be more effective. A novel agent that appears to have substantial activity in the salvage setting is brentuximab vedotin, an antibody-drug immunoconjugate targeting CD30 on the surface of the tumor cells (25). Once the agent binds to CD30, its linker releases the antitubulin drug, monomethyl auristatin E, into the cell and response rates are as high as 70%. One has considerable freedom of choice in selecting the conventional dose regimen. The goal is to achieve as close to a complete response as possible before embarking on the high-dose chemotherapy regimen. Patients entering the high-dose therapy phase with the lowest tumor bulk generally have the highest likelihood of attaining a durable remission.
A number of myeloablative regimens have been employed in the salvage treatment of Hodgkin’s disease including BEAM (26), CVB (27), and high-dose melphalan (28) (see Chapter 38). One has not been convincingly shown to be better than the others. It is important that the treating physician has experience using the regimens at these high doses. Doses and schedules of all the regimens are provided in Table 32-6. Low-dose preparative regimens are also being tested.

Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


