Histone Deacetylase Inhibitors and Demethylating Agents
Steven D. Gore
Stephen B. Baylin
James G. Herman
INTRODUCTION
The past decade has seen an explosive growth, especially at a genome-wide level, in our understanding of the role of chromatin in the normal regulation of gene expression and in the concept of the epigenome.1,2,3 Concomitant with these advances has been the increasing appreciation of the role of epigenetic abnormalities in the progression of cancer4,5,6,7 and the concept of the cancer epigenome. The translational consequences of this research include the possibilities for developing therapies in cancer that target epigenetic abnormalities. These are being explored in clinical trials and several have entered clinical practice.4,5,7,8 Of these epigenetic abnormalities, the most thoroughly examined is the occurrence of abnormal cytosine guanine (CpG) promoter region DNA methylation and associated altered chromatin involving histone modifications, in the transcriptional silencing of genes, including a group of well-defined tumor suppressor genes.4,5,7,8 However, targeting epigenetic processes to downregulate the action of overexpressed genes is also an emerging area of research.9,10 This chapter describes the basis of epigenetic changes in cancer and discusses some of the latest approaches that target epigenetic abnormalities in cancer,11 including those designed to induce the reexpression of silenced genes, for cancer therapy. The two approaches most mature in development are the inhibition of DNA methyltransferases, which mediate the abnormal promoter DNA methylation, and the inhibition of histone deacetylases, which remove histone modifications associated with active chromatin that alone, or in association with DNA methylation, are associated with transcriptional repression.4,5,7,8 However, several exciting newer approaches are now in clinical trials and these will be mentioned.
Aberrant gene function and altered patterns of gene expression are key features of cancer.4 Although genetic alterations remain the best characterized in the development and progression of cancer, increasingly it is appreciated that epigenetic abnormalities cooperate with genetic alterations in multiple ways to cause dysfunction of key regulatory pathways. Through genomic approaches to mutation discovery, there is growing recognition of the frequency of mutations in genes encoding for proteins that regulate the epigenome.12 This chapter will outline the understanding of how each of these epigenetic alterations contribute to cancer and how derivation of therapeutic approaches may depend on understanding the biology of these changes.
EPIGENETIC ABNORMALITIES AND GENE EXPRESSION CHANGES IN CANCER
Epigenetic changes are defined as heritable alterations of gene expression patterns and cell phenotypes, which are not accompanied by changes in DNA sequence.13 This definition clearly delineates the two key features of epigenetic regulation important for an understanding of therapies described in this chapter. Specifically, in contrast to genetic alterations (point mutations, deletions, or translocations), epigenetic changes do not alter the coding sequence of targeted genes. Thus, reversal of epigenetic changes can potentially restore the normal function of affected genes and their encoded proteins. Second, the heritable nature of epigenetic changes—that is, the ability of a cell to pass on regulation of gene expression through DNA replication—suggests that such changes, while relatively stable, can be reversed. Thus, therapeutic reprogramming of patterns of gene expression could theoretically result in a long-term change in the cancer cell phenotype, even after the inducing drugs are removed, although to date, this has not been accomplished.
The fundamental unit that determines epigenetic states is the nucleosome that contains an octamer of histone proteins around which approximately 160 base pairs of DNA are wrapped.13 It is the positioning of these structures, and the three-dimensional aspects of their spacing, and the regulation of this process by posttranslational modifications of the constituent histones that underpins the functions of the epigenome.13,14
Abnormal Gene Silencing
One key alteration in cancer, which can be associated with altered epigenetic control, is abnormal gene silencing. Normally, such silencing is fundamental and required at the level of chromatin and DNA methylation regulation for the life of multicellular eukaryotic organisms. The silencing is critical for regulating important biologic processes, including all aspects of development, differentiation, imprinting, and silencing of large chromosomal domains, including the X chromosome of female mammals.13 For example, the diversity of structure and function of cells derived from epithelial or mesenchymal origin, ultimately differentiating into cells lining the intestine or lung or forming mature granulocytes and myocytes, result from heritable changes in gene expression that are not the result of a change in DNA sequence. Although in many species, silencing can be initiated and maintained solely by processes involving the covalent modifications of histones and other chromatin components, vertebrates utilize an additional layer of gene regulation. This process involves the only natural covalent modification of DNA in humans and is characterized by DNA cytosine methylation that occurs nearly exclusively at the fifth position of the cytosine ring in cytosines preceding guanine, the socalled CpG dinucleotide (Fig. 23.1).13,15
Like most biologic processes, the normal patterns of silencing can be altered, resulting in the development of disease states. Thus, activation of genes normally not expressed, or silencing of a gene that should be expressed, can contribute to the dysregulation of gene function that characterizes cancer and, when stably present, represent epigenetic alterations.4,5,6,7 Most studies have focused on the silencing of normally expressed genes. For the purposes of understanding the rationale behind epigenetic therapy, it is important to understand the mechanisms through which such silencing occurs. Alterations in gene expression associated with epigenetic changes that give rise to a growth advantage would be expected to be selected for in the host tissue, leading to progressive dysregulated
growth of the tumor. Such dysregulation is commonly associated with increases in promoter region DNA methylation and is associated with repressive chromatin changes.
growth of the tumor. Such dysregulation is commonly associated with increases in promoter region DNA methylation and is associated with repressive chromatin changes.
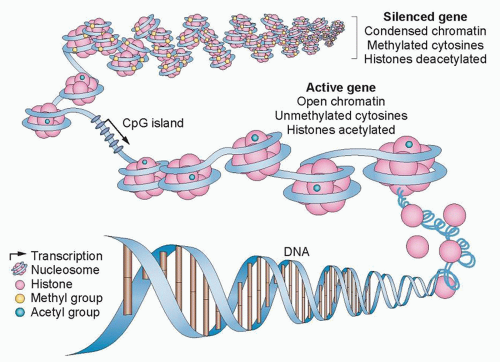 Figure 23.1 Epigenetic regulation of gene expression. In the promoter region, gene expression is controlled by a combination of DNA methylation and chromatin configuration. In normal cells, gene expression is silenced by condensing chromatin, methylating DNA, and deacetylating histones. By contrast, active genes are those with open nucleosome spacing around the transcription start site, are unmethylated, and are associated with acetylated histones. In cancer cells, CpG islands that are rich in cytosine and guanine—and are typically unmethylated to promote gene expression—can be epigenetically silenced by hypermethylation. (Redrawn with permission from Azad N, Zahnow CA, Rudin CM, et al. The future of epigenetic therapy in solid tumours—lessons from the past. Nat Rev Clin Oncol 2013;10:256-266.) |
Changes in DNA Methylation
The importance of abnormal cytosine methylation and gene silencing has been clearly established in the past 2 decades and been shown convincingly to be involved in cancer development.4,5,6,7 The CpG dinucleotide, usually underrepresented in the genome, is clustered in the promoter regions of approximately 50% of human genes in regions termed CpG islands. These regions are largely protected from DNA methylation in normal cells, with the exception of genes on the inactive X chromosome and imprinted genes.16 This protection is critical, because the methylation of promoter region CpG islands is associated with a loss of gene expression.4,5,6,7 Abnormal de novo DNA methylation of gene promoter CpG islands is a very frequent abnormality in virtually all cancer types and is associated with a process that can serve as an alternative mechanism for loss of tumor suppressor gene function.4,5,6,7 Although a limited number of classic tumor suppressor genes can be affected by this process, a patient’s individual cancer may harbor hundreds of such genes.4,5,6,7 Which of these latter genes are drivers of cancer, individually or in groups, versus those which are passengers reflecting only the widespread effects of a global epigenetic abnormality is a leading question in the field and the target of much research.5,6 A clue to the importance of at least groups of the previous DNA hypermethylated genes may come from the fact that an inordinate number of them are involved in holding normal embryonic and adult stem cells in the self-renewal state and/or rendering such cells refractory to differentiation cues.17,18 Normally, these genes are then in a poised expression state and can be induced to be activated or repressed as needed for changes in cell state.18 Abnormal promoter DNA methylation of such genes renders them more repressed and could be a factor in the fact that cancers inevitably exhibit cell populations with enhanced self-renewal or refractoriness to full differentiation.18
Recent studies have also suggested that DNA regions other than promoter CpG islands may undergo changes of DNA methylation in cancer. For example, non-CpG-rich sequences surrounding promoter CpG islands, termed CpG island shores, are abnormally methylated in cancers19 and may be altered in stem cell populations.20 Thus, the relative cancer specificity of changes of DNA methylation in multiple CpG regions makes reversal of these changes by targeting DNA methyltransferases, the enzymes that catalyze DNA methylation, logical for cancer therapeutics.
As a key example of the previous points, perhaps the most studied tumor suppressor gene for promoter hypermethylation is the p16 gene, currently designated CDKN2A, a cyclin-dependent kinase inhibitor that functions in the regulation of the phosphorylation of the Rb protein. Hypermethylation associated with loss of expression of the CDKN2A gene has been found to be one of the most frequent alterations in neoplasia being common in the lung, head and neck, gliomas, colorectal, and breast carcinomas21,22 and other cancer types. A member of the same gene family, p15 or CDKN2B, also regulates Rb and is silenced in association with promoter methylation in many forms of leukemia and in the chronic myeloid neoplasm myelodysplastic syndrome (MDS).23 These two previous changes are of much relevance for the clinical uses of epigenetic therapies discussed later.
As mentioned, many hundreds of genes may be inactivated in a single cancer by promoter methylation,5,6,18,24 providing potential targets for gene reactivation using epigenetic therapies.25,26,27 The latter represents one of the potential ways in which epigenetic therapy may be effective: Multiple genes and gene pathways, all
repressed by changes in DNA methylation and chromatin modification, can be reactivated by DNA methyltransferase inhibitors and histone deacetylase (HDAC) inhibitors (HDACi), thereby restoring normal cell cycle control, differentiation, and apoptotic signaling (Fig. 23.2).8,26,28 In general, methylated CpG islands are not capable of the initiation of transcription unless the methylation signal can be overridden by alterations in factors that modulate chromatin, such as the removal of methylated cytosinebinding proteins. However, reversal of DNA methylation with secondary changes in histone modification or directed reversal of repressive histone modifications represent a target for epigenetic therapies.8,26,28
repressed by changes in DNA methylation and chromatin modification, can be reactivated by DNA methyltransferase inhibitors and histone deacetylase (HDAC) inhibitors (HDACi), thereby restoring normal cell cycle control, differentiation, and apoptotic signaling (Fig. 23.2).8,26,28 In general, methylated CpG islands are not capable of the initiation of transcription unless the methylation signal can be overridden by alterations in factors that modulate chromatin, such as the removal of methylated cytosinebinding proteins. However, reversal of DNA methylation with secondary changes in histone modification or directed reversal of repressive histone modifications represent a target for epigenetic therapies.8,26,28
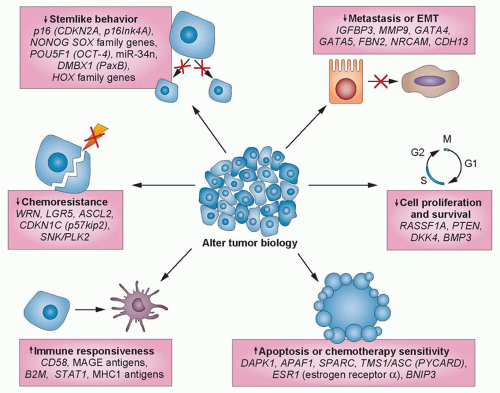 Figure 23.2 Concurrent widespread changes in gene expression with epigenetic therapy. Anticancer efficacy of treatment with epigenetic-modulating agents is associated with extensive changes in gene expression that influence several biologic processes. Gene expression is increased through the direct reversal of epigenetic modifications of genomic DNA, whereas for cancer-promoting genes, gene expression is reduced by the regression of their regulatory genes. EMT, epithelial-membrane transition. (Redrawn with permission from Azad N, Zahnow CA, Rudin CM, et al. The future of epigenetic therapy in solid tumours—lessons from the past. Nat Rev Clin Oncol 2013;10:256-266.) |
Most studies of DNA methylation, particularly in the study of cancer, have focused on CpG island promoter methylation. However, about 40% of human genes do not contain bona fide CpG islands in their promoters.29 The primary focus on CpG islands has resulted from the clear demonstration that CpG-island promoter methylation permanently silences genes both physiologically and pathologically in mammalian cells. However, recent work has shown correlations between tissue-specific expression and methylation of non-CpG islands, including, for example, the maspin gene,30 and as mentioned previously, regions near CpG islands,19,20 suggesting that many additional genes could be regulated, either normally or abnormally, by changes in DNA methylation.
An exciting new area of DNA methylation research involves the role of this change in regulating gene enhancers: small DNA regions that regulate the expression of multiple target genes.31,32,33 The presence of DNA methylation in these areas, which can reside considerable distances from the genes that are being regulated, generally works together with histone modifications to mediate a repressive state for that enhancer.31,32,33 The status of enhancers is also emerging as important for cancer risk states.34
Chromatin in Gene Regulation
Heritable gene silencing involves the interplay between DNA methylation and histone covalent modifications. Complexes of proteins that can regulate how nucleosomes are positioned perform nucleosomal remodeling.35,36,37 What was initially termed the histone code, with reference to how histones are modified, has emerged to be much more complex than originally envisioned. An explosion of research findings during the last several years now allows for an appreciation of how the epigenome is controlled by a complex interplay between a myriad of posttranslational histone modifications that occur on key amino acid residues of these proteins.37 Acetylation, deacetylation, methylation, phosphorylation, and other modifications all modify chromatin structure and thereby alter gene expression.38 Some of the enzymes that catalyze these modifications include HDACs, histone methyltransferases (HMT), and most recently, histone demethylases.13,14,39,40 These modifications help establish heritable states at the start site of genes, but also at enhancers and other transcribed DNA regions not encoding for canonical genes. The latter areas contain noncoding RNAs (ncRNAs) and micro-RNAs (miRNAs), which play key modulatory roles for overall gene expression and protein patterns that can be altered in cancer.41,42,43 Again, much research is being
focused on epigenetic changes in these DNA regions, which may be important to cancer development and, potentially, to cancer management.
focused on epigenetic changes in these DNA regions, which may be important to cancer development and, potentially, to cancer management.
A link between covalent histone modifications and DNA methylation has been clearly established.44,45,46 In this interaction, cytosine methylation attracts methylated DNA-binding proteins and HDACs to methylated CpG sites during chromatin compaction and gene silencing.46,47 In addition, the DNA methylation binding protein (MBD2) interacts with the nucleosomal remodeling complex (NuRD) and directs the complex to methylated DNA.48 This complex also binds HDACs and has recently been identified as a central player for the abnormal silencing of genes associated with promoter DNA hypermethylation in cancer.47 Thus, the three processes of DNA cytosine methylation, histone modification, and nucleosomal remodeling are intimately linked, and alterations in these processes can result in abnormalities of gene expression in cancer-relevant genes.
Enzymes Regulating DNA Methylation and Histone Acetylation
DNA methylation involves the covalent addition of a methyl group to the 5′ position of cytosine. In mammals, three enzymes have been shown to catalyze this transfer of a methyl group from the methyl donor S-adenosylmethionine. Most of the methyltransferase activity present in differentiated cells is derived from the expression of DNMT1.49 This enzyme is thought to be most important in maintaining DNA methylation patterns following DNA replication and thus is referred to as a maintenance methyltransferase. However, the enzyme does possess the ability to methylate previously unmethylated DNA sequences (de novo activity).50 In contrast, the other enzymes, DNMT3a and DNMT3b, are efficient at methylating previously unmethylated DNA and thus are considered de novo methyltransferases. Each of these enzymes possesses a similar catalytic site,51 a fact important for the inhibition of DNMT enzymes by nucleoside analogs, discussed later in this chapter.
DNA methylation is closely associated with changes in the histone modifications. As previously discussed, histone proteins are the central components of the nucleosome, and modifications of the histone tails of core histones are associated with active or repressed chromatin.52 Although it is beyond the scope of this chapter to fully discuss the complex series of modifications to the histone tails of histone H3 and H4, a few well-characterized modifications should be mentioned that are relevant to therapies designed to target epigenetic abnormalities in cancer. In reference to currently investigated epigenetic therapies, changes in histone acetylation are of importance. Acetylation of histones H3 and H4 at key amino acids is associated with the active chromatin present at the promoters of transcribed genes, whereas the absence of histone acetylation is associated with repressed, silenced genes.13,14,53 Histone acetyltransferases (HAT) HDACs have opposing functions to maintain the proper level of histone acetylation for gene expression.13,14,53 HDACs specifically deacetylate the lysine residues of the histone tails, and this deacetylation is associated with condensation of nucleosome positions in what is termed a closed chromatin formation. This scenario is key to transcriptional repression. There are four classes of HDACs.53 Class I HDACs are characterized by their similarity to the yeast Rpd3 HDAC. In humans, this class of enzymes includes HDAC1, -2, -3, and -8. These HDACs are thought to be ubiquitously expressed in tissue throughout the body. In contrast, class II HDACs are similar to yeast Hda1 and include HDAC4, -5, -6, -7, -9, and -10, and they have a greater degree of tissue specificity. Class III HDACs are similar to yeast Sir2 and are set apart from the other classes by their dependence on nicotinamide adenine dinucleotide (NAD+) as a cofactor. Finally, class IV includes HDAC11.53
Of the previously listed HDACs, class I and 2 HDACs have been most closely tied to gene silencing associated with abnormal promoter DNA hypermethylation.48 These are bound to the nucleosome remodeling complex, NuRD.48,49 Experimental decreases in NURD, after use of a DNA demethylating agent, can augment reactivation of many abnormally silenced and DNA hypermethylated genes in colon cancer cells.48 Manipulation of these HDACs is under study in clinical trials, with and without the use of DNA methyltransferase inhibitors, and is discussed later. Another HDAC, SIRT1 in the class III of these proteins, is also involved with gene silencing.54,55 This deacetylase has been linked to silencing of DNA hypermethylated genes, and blocking its activity can be associated with reactivation of such genes.55
Reversal of Layers of Gene Silencing
The interaction between DNA methylation and HDAC activity and repressive chromatin marks in maintaining aberrant silencing of hypermethylated genes in cancer has therapeutic implications for epigenetic therapies. Experimental evidence suggests that DNA methylation functions as a dominant event that stably establishes transcriptional repression. Inhibition of HDAC activity alone, by potent and specific HDACis, does not generally result in the reactivation of aberrantly silenced and densely hypermethylated genes in tumor cells.56 In contrast, treatment with HDACis can reactivate densely silenced genes if the cells are first treated with demethylating drugs, such as 5-azacitidine.56 The clinical implications of this observation are discussed in more detail in the following section (Table 23.1).
DNA Methyltransferase Inhibitors
Originally synthesized as cytotoxic antimetabolite drugs in the 1960s,57 azacytosine nucleosides were recognized as inhibitors of DNA methylation in the early 1980s. The inhibitors 5-azacitidine (5AC) and 2′-deoxy-5-azacytidine induced muscle, fat, and chondrocyte differentiation in mouse embryo cells, in association with a reversal of DNA methylation.58,59 The incorporation of azacytosine nucleosides into DNA in lieu of cytosine residues was shown to be associated with inhibition of DNMT activity.59,60 DNMT inhibition requires the incorporation of decitabine triphosphate into DNA. The incorporated azacytosine nucleoside forms an irreversible inactive adduct with DNMT. The sequential reversal of DNA methylation then results when DNA replication proceeds in the absence of active DNMT.61 The inhibitor 5AC must be phosphorylated and converted to decitabine diphosphate by ribonucleotide reductase before it can be activated through triphosphorylation, whereas decitabine does not require ribonucleotide reductase. The inhibitor 5AC can also be incorporated into RNA. DNMT2, a misnamed protein that is actually an RNA-specific methyltransferase,62 becomes inhibited, leading to the depletion of methylated tRNA.60 This may contribute to the inhibition of protein synthesis and is a potential difference between azacitidine and decitabine.63 The previous DNA methyltransferase inhibitors not only block the catalytic activities of DNMTs, but also trigger degradation of these proteins, especially DNMTs 1 and 3B.64,65,66,67,68 This latter activity is potentially important for their activities for gene reexpression because each of these two proteins, experimentally, possess transcriptional repression properties independent of their DNA methylation catalytic sites.69,70
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


