TABLE 25.1 Classification of Histiocytic Disorders | ||||||
|---|---|---|---|---|---|---|
|
TABLE 25.2 Immunophenotypes of Histiocytic Disorders | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
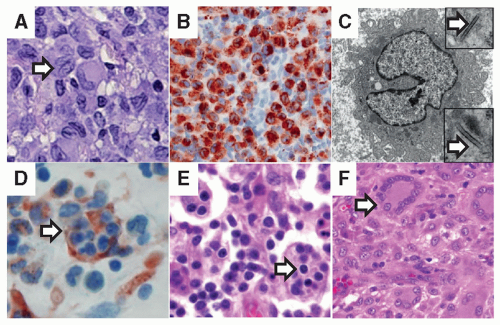 Figure 25.1 Histologic features of histiocytic disorders. A: H&E staining of a typical LCH lesion with characteristic histiocyte with reniform nucleus (arrow) and eosinophilic cytoplasm along with background inflammatory infiltrate (original magnification 400×). B: Langerin (CD207) immunostaining of pathologic dendritic cells within inflammatory lesions is diagnostic for LCH (original magnification 400×). C: Birbeck granules are identified by electron microscopy (arrows) in LCH lesions, as well as in normal epidermal Langerhans cells. This electron micrograph is typical for an LCH lesion with a Langerhans cell histiocyte with abundant cytoplasm and reniform nucleus (original magnification 7,000×; insets 20,000×). D: H&E staining of bone marrow aspirate demonstrates hemophagocytosis (arrow) in a patient with HLH. E: H&E staining of a typical lymph node biopsy from a patient with Rosai-Dorfman disease. The arrow highlights emperipolesis, where viable lymphocytes traffic through histiocytes (original magnification 400×). F: H&E staining of soft tissue juvenile xanthogranuloma with characteristic Touton giant cells (arrow) with nuclei arranged in a wreath-like pattern. Histiocytic cells are seen in the background (original magnification 400×). |
cortical thymocytes, and interdigitating DCs within the dermis and lymph nodes. LCH lesion DCs demonstrate a membranous-to-cytoplasmic pattern with CD1a. LCH DCs may also express langerin (CD207), a type II transmembrane protein. Langerin is normally located on the cell surface and is associated with the formation of characteristic intracytoplasmic Birbeck granules (identified by electron microscopy) thought to be involved in antigen processing. Langerin/Birbeck granules were initially considered specific to epidermal LCs, but CD207 has more recently been identified in other DC lineages.13 Expression of langerin is likely a later event in LCH DC differentiation as many studies describe variable expression of CD207+ among CD1a+ and BRAF-V600E+ histiocytes within LCH lesions (reviewed in Ref. 14).
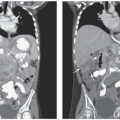
 Figure 25.2 Clinical features of histiocytic disorders. A: Cutaneous Langerhans cell histiocytosis. An erythematous, papular rash resembling a Candida diaper rash. B: Skull is the most common site for LCH lesions in children. They can be isolated, part of multifocal bone disease, or part of multisystem disease. C: Lung lesions may arise as part of multisystem LCH, or in adults as isolated disease generally in the context of cigarette smoking. The computed tomographic (CT) image demonstrates typical cystic disease resulting from LCH lung lesions. D: Multisystem LCH can present with hepatosplenomegaly, as demonstrated by the MRI image. E: Neurodegenerative disease may be observed along with or following initial presentation of LCH. This MRI image illustrates the characteristic T2-weighted or FLAIR image with hyperintense signal in the cerebellum. |
with marrow hemophagocytosis that mimics hemophagocytic lymphohistiocytosis.24
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


