The ‘familial hypercholesterolemia phenotype’ has clinical and laboratory features usually ascribed to the heterozygous form of familial hypercholesterolemia (FH) (Table 1.1) secondary to a mutation of the low-density lipoprotein (LDL) receptor gene (LDLR). Over time, this phenotype has been observed, albeit more rarely and often incompletely, in conditions with different etiologies, some inherited in a dominant fashion (i.e. homozygotes do not have a more severe phenotype than the heterozygote) and others as a recessive trait. They essentially mimic heterozygous FH, which is a codominantly inherited disease (i.e. homozygotes have a more severe phenotype than heterozygotes). These disorders are monogenic and include familial defective apolipoprotein B100 (FDB) (APOB gene), autosomal dominant hypercholesterolemia (PCSK9), cholesterol 7α-hydroxylase deficiency (CYP7A1), familial sitosterolemia (ABCG5 or ABCG8) and autosomal recessive hypercholesterolemia (ARH) (ARH). From this degree of heterogeneity, it is anticipated that other gene defects will also account for this phenotype. Autosomal dominant hypercholesterolemia (ADH) has been referred to as comprising ‘FH1’, the classical FH, ‘FH2’ (FDB) and ‘FH3’, attributed to a PCSK9 defect. As discussed below, there are variations of the typical FH phenotype that might not fully justify this classification, such as dominance versus co-dominance, variable expression and less severe manifestations.
Dominant monogenic forms
Familial hypercholesterolemia
The classical nosological entity called familial hypercholesterolemia had been part of the medical literature for a long time before its mechanism was fully unraveled by the seminal work of the 1985 Nobel laureates, Joseph L Goldstein and Michael S Brown. For a monogenic disorder, it is relatively common, its frequency varying from 1 in 500 in most parts of the world to as much as 1 in 80 in regions where a founder effect exists due to factors such as endogamy, consanguinity, geographic isolation and limited genetic admixture. Such regions include, among others, Lebanon, Finland, South Africa (Afrikaners), Canada (French-Canadians) (1.1) and Israel (FH-Sephardic and FH-Lithuania). Its importance stems also from the devastating severe and premature cardiovascular consequences for the affected members (50%) of a family, stressing the need for early diagnosis. As there is treatment that will prevent or markedly delay the complications of this condition, early intervention may result in a normal life. In other words, it is a treatable killer and worldwide efforts have therefore been made to identify individuals and families at risk. MEDPED, ‘make an early diagnosis, prevent an early death’ is one such initiative (www.cholesterol.med.utah.edu/medped/). In certain communities, it may be a major health problem, disabling or killing many young adults.
Table 1.1 Characteristics of familial hypercholesterolemia (FH)*
*These characterize the ‘FH phenotype’ and it is helpful if the family history also reveals the presence of tendon xanthomas or the presence of a homozygote.
The etiology of FH is well established. The metabolic defect (1.2), which leads to a large increase in plasma levels of LDL-cholesterol (LDL-C) (two to three times that of normal subjects), is attributed to mutations of the LDL receptor gene that result in a decreased number or total absence of LDL receptors or, alternatively, in expression of dysfunctional receptors. This ubiquitous receptor allows uptake and degradation in the liver of LDL, the major lipoprotein carrier of circulating cholesterol (mostly cholesteryl esters), allowing excretion of the latter into the bile (as free cholesterol). It is essential for bringing cholesterol, a major constituent of membranes, to cells. The defect causes a considerable delay of LDL clearance from plasma. Normal subjects catabolize about 45% of their LDL pool per day, whereas the fractional catabolic rate is 25-30% for heterozygotes and 15% for homozygotes. There is also a delayed clearance of intermediate-density lipoproteins (IDL), which represent ‘remnant’ lipoproteins and an increased conversion of IDL into LDL. Recent studies by Tremblay and co-workers have shown that there is also a 50% increased production rate of verylowdensity lipoprotein (VLDL) apolipoprotein B (apoB) – 100 in heterozygotes and 109% in homozygotes. This finding sheds light on an old debate. The LDL particles are large and buoyant in this condition because there is excess cholesterol associated with apoB, the main apolipoprotein associated with these cholesterol-rich particles. There are over 800 mutations of the LDL receptor gene (LDLR), which is located on the short arm of chromosome 19 (19p13.1-13.3) (1.2). They have been indexed continually in the UK since October 1996 on a dedicated website (www.ucl.ac.uk/fh/) and in France since April 1998 (www.umd.necker.fr/). A classification of the various defects of the LDLR gene has also been established (Hobbs et al. 1990). The abnormality may involve synthesis in the endoplasmic reticulum, transport of newly synthesized receptors to the Golgi complex, transport to the cell surface, clustering in the surface-coated pits or binding affinity for LDL, depending on the portion of the receptor gene that is affected (1.1). For practical purposes, it is useful to know if the receptor activity is impaired (receptor defective) or not functional at all (receptor negative). Severity and resistance to treatment are greater in the latter. This is especially important for homozygotes and can be determined by identifying the mutation or testing LDL receptor activity in cultured skin fibroblasts or blood mononuclear cells.
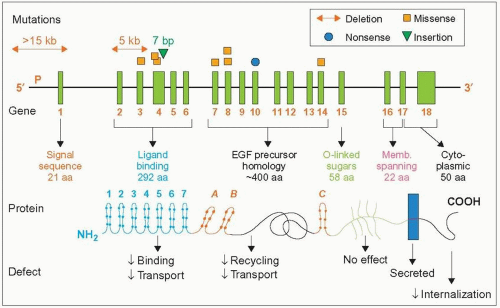
1.1 French-Canadian mutations of the LDL receptor gene. This figure provides information on the type of mutations (those most frequent in the Province of Quebec), the structure of the gene, what the exons (green vertical bars) code for, the structure of the protein (domains and number of amino acids), and what abnormalities might be expected when one such domain is affected by the mutation. The dots on the protein sequence represent cysteines. The mutations represented are reported in the following papers: Hobbs HH et al. (1987). N Engl J Med, 217: 734; Leitersdorf E et al. (1990). J Clin Invest, 85: 1014; Ma Y et al. (1989). Clin Genet, 36: 219; Simard J et al. (1994). Hum Mol Genet, 3: 1689; Assouline L et al. (1995). Pediatrics, 96: 239; Couture P et al. (1998). Hum Mutat, 1: S226.
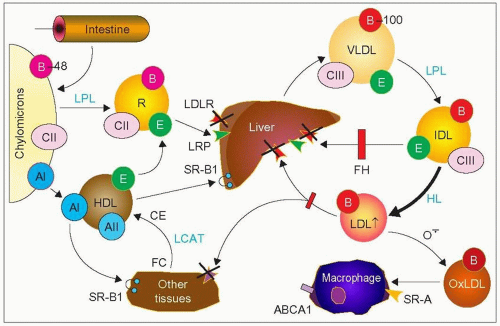
1.2 Metabolic defects in familial hypercholesterolemia (FH). Abnormalities of the low-density lipoprotein (LDL) receptor (LDLR, small red arrowheads marked with a cross) prevent the normal clearance of cholesteryl esters (orange in the circles) transported by LDL, which increase in plasma. Characteristically, these LDL particles transport as the main protein apolipoprotein B-100 (apoB-100) on their surface (red circles) that interact with the LDL receptor for uptake by the liver allowing eventual excretion of cholesterol into the bile. ApoB-100 is carried by very-low-density lipoproteins (VLDL) secreted by the liver, and their degradation products, including intermediate-density lipoproteins (IDL). This degradation takes place gradually by delipidation and loss of surface proteins such as apoCIII and apoE. Lipoprotein lipase (LPL) allows transformation of VLDL into LDL and hepatic lipase (HL), that of IDL into LDL.
In FH, IDL are still taken up by the remnant receptor, LDL receptor-related protein (LRP), but this pathway is limited (red bar) by the LDLR defect. On the other hand, more IDL are transformed into LDL (large black arrow). Some LDL particles, because of the prolonged residence time, may become oxidized (OxLDL), taken up by macrophages via various scavenger receptors (SR-A) and contribute to the formation of foam cells present in xanthomas and atheroma. ApoB-48, a smaller apoB formed by splicing of the APOB gene mRNA present on chylomicrons is not taken up by the LDL receptors but by another dedicated receptor. Breakdown delipidated products of chylomicrons become remnants (R) as they gain apoE which can interact with LRP for uptake by the liver. There is evidence from stable isotope studies that the high LDL levels in FH may also be due to some increase in VLDLapoB production rate relative to normal subjects. When one refers to apoB without the -100 or the -48, it is usually taken to be apoB-100 or, alternatively, all forms of apoB. SR-B1 refers to the scavenger receptor class B type 1 (also called Cla-1). It is a receptor that allows transfer of cholesteryl esters (CE) from high-density lipoproteins (HDL) to different tissues as well as transfer of cholesterol from tissues to HDL. Lecithin:cholesterol acyl transferase (LCAT) is an enzyme that esterifies free cholesterol to CE during HDL remodelling from discoid to spherical and mature HDL.
The diagnosis of heterozygous FH (Table 1.1) is based essentially on:
the family history (premature atherosclerosis and early deaths especially in males, tendon xanthomas, presence of a homozygote)
premature atherosclerosis (a myocardial infarction may occur as early as in the third or fourth decade)
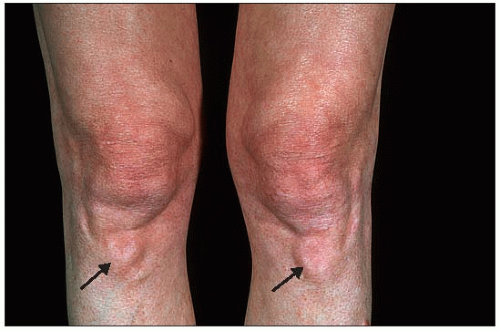
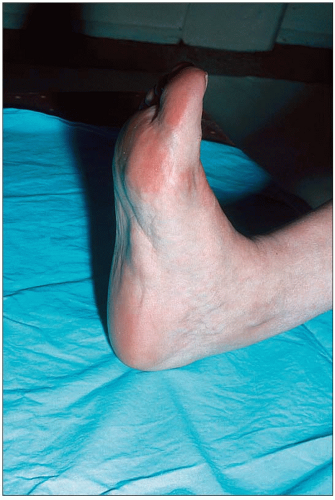
the clinical manifestations of hypercholesterolemia (especially tendon xanthomas – Achilles or extensor tendons of the fingers – but also periosteal, prepatellar, plantar, tricipital and tuberous xanthomas as well as corneal arcus) (1.3,1.4,1.5,1.6,1.7) and atherosclerosis (arterial bruits, angina, intermittent claudication, Leriche syndrome, transient ischemic attacks, etc.)
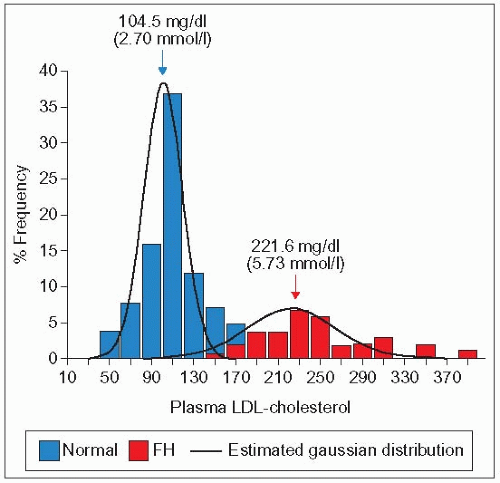
high levels of plasma LDL-C (>95th percentile) (1.8)
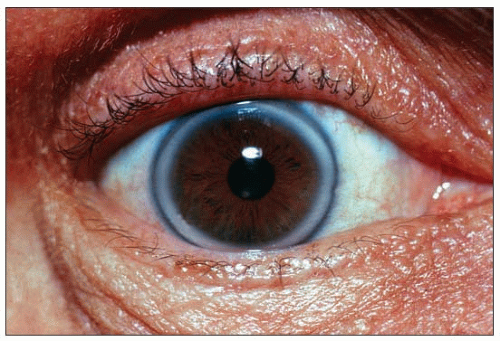
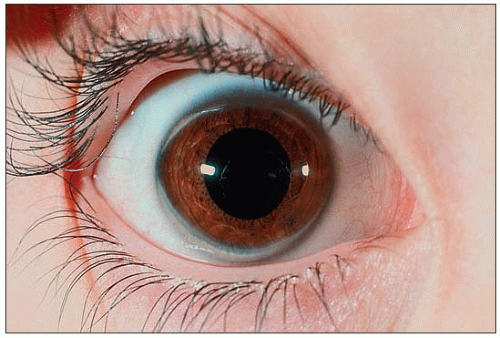
1.3 Corneal ring in a patient with heterozygous familial hypercholesterolemia. A corneal arcus usually starts as a small, barely visible whitish crescent in the upper and/or lower part of the cornea. Careful attention needs to be paid, using good lighting, in order to notice them. They may grow to the point, as seen here, where they form a quite obvious corneal ring. Note the clear space between the ring and the periphery of the cornea. Corneal arcus and rings may be seen in individuals of African origin in the absence of hyperlipidemia.
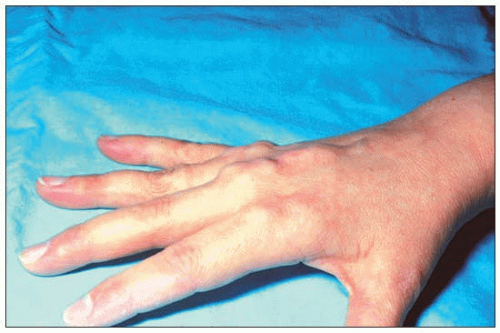
1.4 Extensor tendon xanthomas in familial hypercholesterolemia (FH). The presence of these xanthomas often allows a diagnosis of FH at first sight. They tend to regress readily with major reductions in LDL-C, e.g. with statin therapy. Even when discrete, they are rarely missed. Palpating the extensor tendons when FH is suspected may reveal incipient lesions.
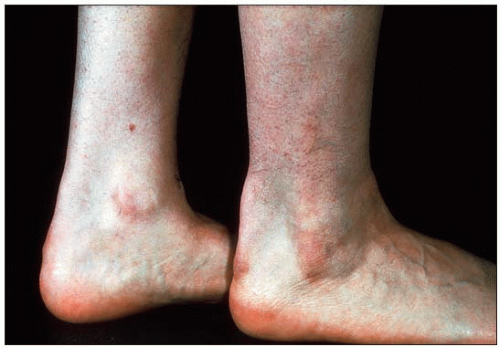
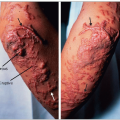
Detection of affected individuals is more difficult in childhood. The family history, combined with a blood sample for LDL-C is most useful in children. In women, manifestations are delayed by about 10-15 years compared with men (1.9). Evolution of tendon xanthomas may be assessed and followed using standardized X-ray techniques (1.10), ultrasonography or magnetic resonance imaging. The xanthoma size correlates with the duration and severity of the disease (1.11). Achilles tendon xanthomas are prone to sporadic inflammation, causing painful acute tendinitis (1.12).
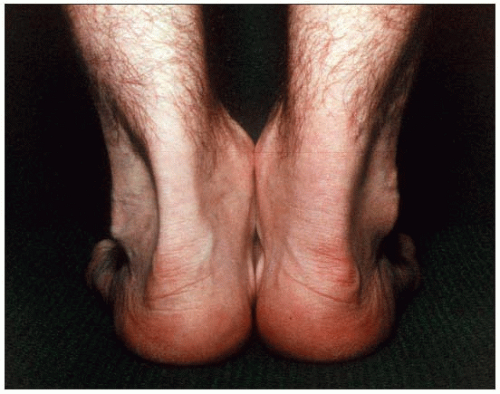
1.5 Lateral view of Achilles tendon xanthomas in familial hypercholesterolemia. This lateral view of tendon xanthomas shows the growth of these lesions anteroposteriorly as well as laterally. These xanthomas are often missed if smaller in size and the physician does not palpate carefully. Pinching this area while sliding the fingers downward will reveal olive-shaped lesions or diffuse thickening of the tendon.
1.6 Periosteal xanthomas of the anterior tuberosity of the tibia. These lesions are not always noticed by the patients themselves as they progress very slowly, occasionally to huge proportions. They may become inflamed and painful or tuberous xanthomas may develop in the same area. Xanthomas often develop at sites of repeated trauma.
1.7 Plantar xanthoma in a woman with heterozygous familial hypercholesterolemia. Plantar xanthomas, like Achilles tendon xanthomas, become fibrotic and hard with time and regress poorly. They can become very debilitating and impair shoe fitting and walking.
Because LDL-C level is the best biochemical marker of FH, the most threatening and most directly linked to the causal defect, it remains the centre of attention. FH was classified in the Fredrickson era among subjects presenting a ‘type IIa’ lipoprotein phenotype (isolated hypercholesterolemia), and ‘type IIa’ became wrongly synonymous with FH. Indeed, the hypercholesterolemia may occasionally be associated with hypertriglyceridemia (becoming Fredrickson’s ‘type IIb’). This associated hypertriglyceridemia may be due to a second gene defect, another medical condition or environmental factors, or may be part of the defect in a particular family (triglyceride-rich remnant lipoproteins are cleared to some extent by the LDL receptor). The diagnosis may be established in most cases without resorting to determination of the genetic defect(s) using molecular biology techniques. This task, when required, is easier in communities where a founder effect exists, because only a few mutations may explain a majority of cases (1.1). When a doubt exists, some specialized lipid clinics may be able to help with identification of the gene defect.
1.8 Bimodal frequency distribution of LDL-C in a large kindred with hypercholesterolemia. This diagram demonstrates the bimodal frequency distribution of low-density lipoprotein (LDL)-cholesterol in a single pedigree segregating for familial hypercholesterolemia (FH) (120 members). The red bars represent the patients in whom a clinical diagnosis of FH was made. The blue bars represent the non-affected members. Note the wide range of LDL-C in the affected subjects and the overlap with the normal population. The mutation of the LDLR segregating in this family was the French-Canadian-1 mutation (deletion >15 kb in the promoter region encompassing exon 1 and preventing expression of the LDL receptor). Reproduced with permission from Davignon J et al. (1991). In: Steiner G, Shafrir E (eds). Primary Hyperlipidemia. McGraw Hill, New York, p. 201.
The differential diagnosis must include other causes of xanthoma tendinosum since they constitute quite a reliable diagnostic criterion; these include lesions that can be mistaken for xanthoma tendinosum (Table 1.2). Few hereditary dyslipidemias apart from heterozygous FH and autosomal dominant hypercholesterolemia (FH3) have such high levels of cholesterol except the other monogenic forms discussed below and familial dysbetalipoproteinemia (type III), but in this condition, LDL-C measured directly is not elevated. Severe familial combined hyperlipidemia with isolated hypercholesterolemia (lipoprotein phenotype IIa) may rarely have LDL-C levels similar to those found in the lower distribution of FH, but tendon xanthomas have not been reported. Atherosclerotic vascular disease in the family tends to occur later in life than in FH and the variation in lipoprotein phenotype in first-degree relatives is typical. Among the secondary forms of hypercholesterolemia the most likely to be confused with FH are primary biliary cirrhosis, nephrotic syndrome and hypothyroidism, all potentially associated with very high levels of LDL-C.
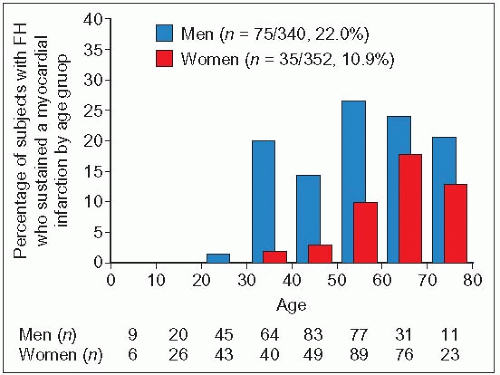
1.9 Myocardial infarction incidence by age group in men and women with heterozygous familial hypercholesterolemia (n = 692). This figure illustrates how myocardial infarction may occur early in life, even in the relatively protected Japanese population. It also shows the delay of 10-15 years (relative to men) before women develop myocardial infarctions. Reproduced with permission from Mabuchi H et al. (1989). Development of coronary heart disease in familial hypercholesterolemia. Circulation, 79: 225-232.
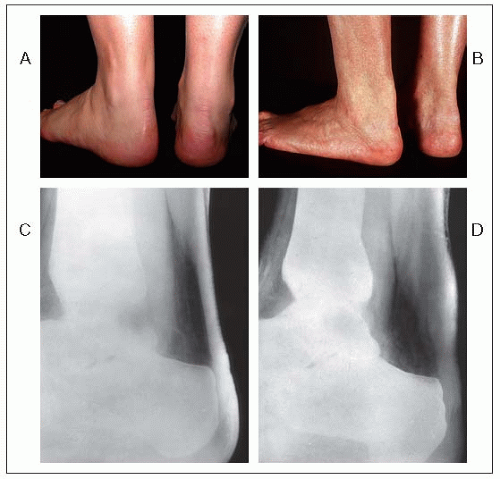
1.10 Normal and xanthomatous Achilles tendons and their radiological assessment. Panels A and C: Achilles tendons of a normal woman. Panels B and D: Nodular thickening of left Achilles tendon in a woman of the same age, heterozygotic for familial hypercholesterolemia. A soft tissue standardized radiological technique was used. The thickening of the Achilles tendon in the anteroposterior dimension is obvious with this radiological technique.
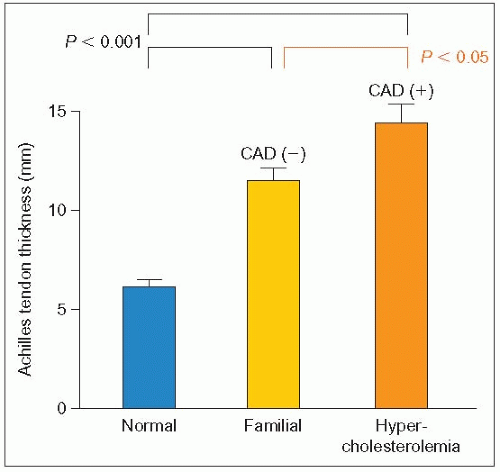
1.11 Tendon xanthoma thickness in FH patients without or with coronary artery disease. This figure shows that the size of Achilles tendon xanthomas is larger in subjects with coronary artery disease (CAD). The severity of the clinical manifestations is a function of the magnitude of the hypercholesterolemia, its duration and the presence of other cardiovascular risk factors. Redrawn from Mabuchi H et al. (1978). Achilles tendon thickness and ischemic heart disease in familial hypercholesterolemia. Metabolism, 27: 1672-1678.
1.12 Tendinitis of xanthomatous right Achilles tendon in a heterozygous familial hypercholesterolemia (FH) patient. Both tendons are xanthomatous, but the right one is enlarged, inflamed and painful. This is not infrequent in patients with FH. In the authors’ experience, it may occur in young men or women particularly responsive to treatment, after a large and rapid decrease in plasma low-density lipoprotein-cholesterol.
Treatment almost always necessitates the addition of a statin (an HMG-CoA reductase inhibitor) to a cholesterollowering diet. Often, large doses of statin are needed and combination therapy with a resin (a bile acid absorption inhibitor) or with ezetimibe (a cholesterol absorption inhibitor) may be necessary. Various forms of LDL apheresis have been used in some very severe heterozygous FH refractory to drug therapy.
The homozygous (inheritance of two identical defective genes) or double heterozygous (two different gene defects at the same locus) forms are extremely rare (approximately 1 in 300 000 to 1 in 1 000 000). The clinical picture is so dramatic that the diagnosis is rarely missed (1.13,1.14,1.15,1.16,1.17). Xanthomas are diverse (tuberous, planar, tendinous, xanthelasma), extensive, ubiquitous (friction sites, elbows, knees, popliteal space, palms, plantar aponeurosis, gluteal crease) and may be present at birth. LDL-C levels may be four to six times the upper limit of normal and the type of mutation may also influence the plasma levels (1.18). Coronary death can occur as early as two years of age and the affected patients, whether male of female, rarely live beyond the third decade. One typical complication, in addition to myocardial ischemia and infarction, is aortic stenosis (1.19), which is sometimes seen in heterozygotes with severe disease. Differential diagnosis must include ARH and a condition reported in the past as pseudo-homozygous hypercholesterolemia (1.20). However, some of these cases may have had an ARH defect, especially when they responded well to dietary or statin treatment. Statins have a modest effect that is enhanced by combination with ezetimibe and LDL apheresis. Probucol, a major antioxidant, now withdrawn from the market but still available in Japan, has been reported to reduce the size of tendon xanthomas in homozygous FH. ‘Last resort’ treatments have been used, including porto-caval shunts, gene therapy and liver transplantation, with all but the latter having little success.
Table 1.2 Differential diagnosis of tendon xanthomas
* Reported by Ng et al. (1994). J Clin Invest, 93: 223.
1.13 Corneal arcus in a 5-year-old with homozygous familial hypercholesterolemia (FH). Upper and lower corneal crescents in 5-year-old with homozygous FH. The lower arcus is unusual in not being separated from the sclera by a clear space as observed in the upper one and with the full corneal ring illustrated in 1.3.
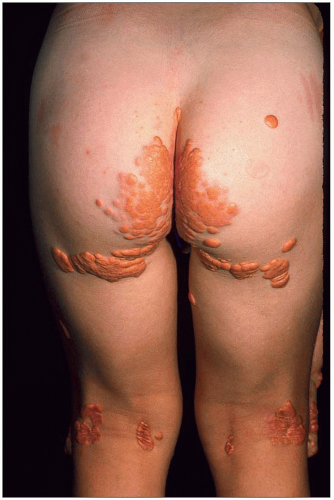
1.14 Raised planar xanthomas in creases and sites of friction in homozygous familial hypercholesterolemia (FH). These raised planar xanthomas in a 5-year-old homozygous FH boy have the typical orange colour and develop at sites of friction between the buttocks and in the popliteal space.
It is worth remembering that in FH, and especially in the homozygous form, the first concern of the doctor should be the accelerated atherosclerosis that accompanies this condition. The autopsy specimen presented in 1.21 showing severe aorto-femoral atheroma and aneurysmal weakening of the wall reminds us of the consequences of failing to intervene early and aggressively in these cases. A sense of urgency should always be uppermost in the mind of the doctor.
Familial defective apolipoprotein B-100
FDB was identified and its etiology determined in 1985-1986 by Grundy and colleagues at the University of Texas in Dallas and Mahley and co-workers at the Gladstone Research Foundation Laboratories in San Francisco. It is an autosomal dominant monogenic disorder due to point mutations in the APOB gene (1.22). This very large gene (43 kb, 29 exons) was mapped to the distal short arm of chromosome 2 (2p23-p24) in 1985-1986 by investigators from several laboratories (including Knotts, Chan, Law, Deeb and their co-workers). These mutations impair the affinity of apoB, the ligand, to its receptor, the LDL receptor (1.23), hence the synonym of ‘familial ligand-defective apoB-100’ (FLDB). Five mutations in exon 26 of APOB may cause this condition, Arg3500→Gln (the first common mutation identified), Arg3500→Trp, Arg3531→Cys, and Arg3480→Trp (Sweden) and Thr3492→Ile (Poland). A sixth recently reported mutation His3543→Tyr is four times more frequent than the R3500Q variant (for amino acid nomenclature see www.chem.qmul. ac.uk/iupac/AminoAcid/AA1n2.html#AA1). It appears to be associated with a variable but moderate degree of LDL-C elevation and a reduced apoB-100 fractional catabolic rate. An Asn5316→Lys mutation of APOB has little impact on the lipoprotein profile but changes apoB conformation. FDB is inherited as an autosomal dominant trait with incomplete penetrance or variable phenotypic expression.
The prevalence of FDB varies widely from country to country. In Caucasians from the USA and Europe, it averages 1 in 500 to 1 in 700. It is high in Switzerland (1 in 209 to 1 in 230) and Poland (1 in 250) and rare in Mediterranean countries. It has not been found at all in the Turkish or Finnish populations, or among hypercholesterolemic Japanese or Israelis. From prevalence and haplotype studies, Miserez and Muller at the Basel University Clinics speculated that the common mutation originated from Celtic ancestors in a region between Lake Geneva, the Jura mountains and the Rhine (in the northwestern part of Switzerland the prevalence of this mutation is 1 in 114), perhaps as early as the Mesolithic period (6000-10 000 years ago) (1.24). This hypothesis is consistent with a previous study from Myant and colleagues who used a combined molecular and population genetic approach to estimate the age of the mutation to be 6000-7000 years.
The impaired ligand-receptor interaction (20-30% of normal binding to fibroblasts) results in delayed clearance of defective LDL particles with a residence time of LDL–apoB 3.6 times longer than that of normolipidemic controls (8.2 vs. 2.3 days). This is associated with decreased production of LDL and enhanced removal of the apoE-containing VLDL, as demonstrated by Schaefer and co-workers in 1997 in a subject homozygous for the common mutation R3500Q (1.25). The residence time of VLDL–apoB is also increased, but that of VLDL-apoE is reduced since apoE becomes the favoured ligand to clear particles via the LDL receptor and LDL receptor-related protein (LRP). In addition, LDL isolated from these subjects has increased susceptibility to oxidation. The molecular mechanism whereby the apoB-100 mutations cause the phenotype was unravelled by Borén and co-workers in 2001. Arginine at residue 3500 is essential for normal receptor binding. The carboxyl terminus of apoB-100 is necessary for mutations affecting this arginine at residue 3500 to disrupt LDL receptor binding. Borén and colleagues drafted a model illustrating that Arg3500 interacts with Trp4369 and facilitates the conformation of apoB-100 required for normal receptor binding of LDL; the carboxyl terminal of apoB-100 interacts with the backbone of apoB-100, which in turn wraps around the LDL particle (1.26).
Only gold members can continue reading. Log In or Register to continue