White
African Americans
Asian American and Pacific Islander
American Indian and Alaska Native
Hispanic/Latino
(a) Incidence
Males
58.2
68.4
44.1
38.1
50
Females
42.6
51.7
33.1
30.7
35.1
(b) Mortality
Males
21.4
31.4
13.8
20
16.1
Females
14.9
21.6
10
13.7
10.7
Twin [2] and family [3–5] studies have shown that genetic risk factors influence CRC incidence in up to 35 % of cases, but within that 35 % the level of risk conferred by genetic factors that predispose to CRC varies enormously. In comparison to the risk for a person with no relatives with CRC (Fig. 25.1), the risk for a person who has a single first-degree relative with CRC is threefold greater (6 % lifetime risk), and it is eightfold greater for a person who has two first-degree relatives with CRC (17 % lifetime risk). Moreover, risk is increased fivefold if the affected first-degree relative was diagnosed before age 45 [6, 7]. There is also increased risk of developing CRC in the first-degree relative of a person who has been diagnosed with one or more adenomatous colonic polyps [8, 9]. These correlations between family history and CRC susceptibility are propelled by a range of genetic mechanisms, from high-risk, disease-causing mutations that ablate gene function to low-risk genetic variants that perturb gene function in subtler ways. In addition, genetic risk factors can interact with each other and with nongenetic factors to increase or decrease CRC risk. However, the impact of interaction on risk is poorly understood.

Fig. 25.1
Familial colorectal cancer risk. Estimates of the absolute lifetime risk of colorectal cancer as a function of family history of colorectal cancer. Adapted from Burt [10].
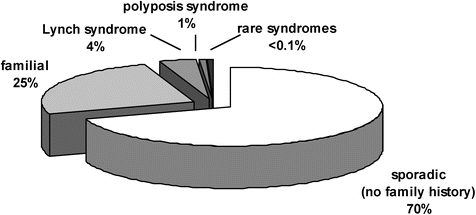
The correlations between family history and CRC risk are also reflected in the types of family histories in which CRC occurs and the proportions of these types. Approximately 70 % of CRCs occur in persons with no known family history of CRC, so-called sporadic CRC (Fig. 25.2). In ~25 % of CRC cases, the person with CRC has up to two first-degree or second-degree relatives with CRC, which is often termed familial CRC [10, 11]. In ~5 % of cases, there is a dramatic family history (three or more CRCs in multiple generations in the family). Cases with strong family histories are often associated with a disease-causing mutation in a known hereditary CRC gene. For example, in ~3 % of all CRC cases, a mutation in one of the mismatch repair (MMR) genes is present. When a disease-causing MMR gene mutation is present, the CRC predisposition syndrome is referred to as Lynch syndrome or hereditary non-polyposis colorectal cancer (HNPCC). In ~1 % of CRC cases, a mutation in the adenomatous polyposis coli (APC) gene is present, which is strongly associated with colonic polyposis. When a mutation in APC is present, the syndrome is referred to as familial adenomatous polyposis (FAP). In <1 % of CRC cases, a mutation is present in a gene that is associated with a hamartomatous syndrome, including familial juvenile polyposis, Cowden’s disease, and Peutz-Jeghers syndrome (PJS).
Given the range of risks associated with known genetic risk factors and the distribution of family histories of CRC, what do we mean by hereditary CRC? Certainly, syndromes such as Lynch syndrome and FAP, which are associated with single-gene mutations and found in multiple family members, are hereditary in the classical Mendelian sense. When a disease-causing mutation is known to be present in a person, the quantitative information regarding cancer risk is available. However, family history is an imprecise tool for predicting the presence of a disease-causing mutation. In the following examples, we present some of the reasons for this lack of predictive power. These examples show how family history can be a poor predictor of CRC risk, and they underline the importance of developing more precise and reliable genetic tools to identify persons at high risk of CRC.
25.1.1 Variable Penetrance
One and the same MMR gene mutation can be found in families with a range of family histories of CRC from very strong to relatively weak. One reason for this range of family histories is that penetrance is variable (penetrance is defined as the probability of developing the disease given the presence of the mutation). On average, penetrance of MMR mutations is less than 50 %. The reasons for variable penetrance are not well understood. Probabilistic events (family size, ascertainment bias, etc.) may explain some of the variation in family history. Deterministic factors also probably play an important role, including genetic factors that modify the risk conferred by the cancer-causing mutation (these genetic factors are called modifiers), exposure to environmental factors, or both.
25.1.2 New Mutations
Up to 30 % of APC mutations are new mutations that are not associated with previous family history of CRC, but which are every bit as dangerous to the present and subsequent generations. Clearly, in these cases, there is no family history to help predict disease occurrence.
25.1.3 Recessive Inheritance
Late-onset Mendelian syndromes associated with increased susceptibility that are caused by recessive genes are difficult to identify and characterize because family sizes are generally small and penetrance is dependent on age. Neither parent is likely to have been diagnosed with CRC and the probability for each son or daughter of having both disease alleles is 25 %. A perfect example of this scenario is the recently described CRC syndrome MYH-associated polyposis (MAP) , which is associated with biallelic recessively inherited mutations in MUTYH (also referred to as MYH). Many more recessive CRC syndromes may explain CRC incidence in persons with no previous family history or in familial CRC.
25.1.4 Polygenic Inheritance
Strong family histories of CRC may not be associated with classical single-gene, Mendelian mutations, but rather with the presence of multiple genetic risk factors that combine to create high risk. This scenario can resemble recessive inheritance in that each parent may be at low risk of CRC because he or she carries one or two low-risk or moderate-risk genetic factors. Segregation of genetic factors to a son or daughter from both parents is thus a relatively low-frequency event.
25.1.5 Hereditary Colorectal Cancer
For the purposes of this chapter, we define hereditary CRC as a syndrome caused by one or more genetic risk factors that are associated with a strong family history of CRC, which can be established statistically through tests of genetic segregation in families. Consequently, the main focus here is on the syndromes defined by disease-causing mutations in single-gene Mendelian disorders. There is a general consensus now that the known single-gene disorders do not account for all the genetic variation that influences CRC susceptibility and that many low-risk or moderate-risk genetic variants need to be identified in order to explain the remaining heritability of CRC. We note that numerous reports describing low-risk CRC susceptibility factors have recently been published and elucidation of the role and impact of the major low-penetrance risk alleles is an active area of current genetic investigation [12]. These low-risk genetic factors explain but a small fraction of the yet-to-be-determined CRC susceptibility factors. Consequently, the search goes on to identify genetic risk factors that explain the heritability of CRC with the hope that these factors will some day allow a more precise and reliable prediction of CRC risk.
25.2 Lynch Syndrome
The non-polyposis disorders are so called to distinguish them from those syndromes that lead to the formation of numerous polyps of the lower gastrointestinal tract. Foremost among the non-polyposis disorders is Lynch syndrome (hereditary non-polyposis colorectal cancer). This syndrome arises from mutations in the MMR pathway and results, to varying degrees, in a phenotype of early-onset CRC and extracolonic malignancies. The MMR genes function in a specific type of DNA repair that ensures high fidelity of DNA replication. When MMR function is deficient in a cell, it is associated with a high mutation rate, which drives carcinogenesis. The high mutation rate can be detected using a simple polymerase chain reaction (PCR) assay that tests for microsatellite instability (MSI) . Tumors that exhibit MSI have the hypermutation phenotype that is associated with loss of MMR gene function.
Historically, the earliest descriptions of Lynch syndrome came from the pathologist Aldred S. Warthin at the University of Michigan who, in 1913, published a review of several cancer-prone families [13]. Approximately 50 years later, Henry T. Lynch and colleagues collected data from these same families and recognized that CRC was the most prominent cancer in this syndrome. Initially, this familial syndrome was given the name hereditary non-polyposis colorectal cancer (HNPCC) to distinguish it from hereditary conditions in which cancers arise in a milieu of carpeting colonic polyps. In 1984, the name Lynch syndrome was proposed in honor of Dr. H.T. Lynch’s seminal observations. Moreover, HNPCC was thought to be a misleading name because polyps are still encountered albeit not to the degree seen in polyposis conditions. Arguably, the definition of polyposis is somewhat arbitrary. In 1993, the field of Lynch syndrome genetics was energized by the identification of MSH2 and, shortly thereafter, MLH1 as genes the mutation of which results in Lynch syndrome. In the 1990s, clinical criteria and guidelines were established including the Amsterdam criteria and Bethesda guidelines that are used to guide molecular genetic testing in individuals who might be affected by the syndrome (Table 25.2). The Amsterdam criteria were devised to enrich for families with mutations to help in the identification of the culprit genes; the Bethesda guidelines were an attempt to devise a more inclusive set of rules to identify mutation carriers. These guidelines nonetheless cannot be used to identify all persons with disease-causing mutations or families in which MMR gene mutations are segregating, because age of disease onset is variable, penetrance is variable, and family sizes are often small. Universal screening—MSI testing, immunohistochemistry (IHC) for MLH1, MSH2, MSH6, and PMS2, or both—on all CRCs has been recommended, which obviates the need for guidelines, and it has slowly gained widespread acceptance. As whole-genome DNA sequencing becomes more efficient and lower cost, we can foresee a day in which genetic screening using whole-genome sequencing would help overcome the limitations of clinical guidelines.
Table 25.2
Amsterdam II criteria and revised Bethesda criteria
Amsterdam II | Revised Bethesda | Patient who meets Amsterdam II criteria |
|---|---|---|
Three or more relatives with a Lynch syndrome- associated cancer | CRC diagnosed in a patient less than 50 years old | |
Two or more successive generations affected | presence of synchronous or metachronous CRC or other LS-associated tumor regardless of age | |
One or more relatives diagnosed before age 50 | CRC with microsatellite instability in patient less than 60 years old | |
One should be a first-degree relative of the other two | CRC diagnosed in one or more first-degree relative with a LS-associated tumor, with one of the cancer diagnosed less than 50 years old | |
Exclude familial adenomatous polyposis | CRC diagnosed in two or more first- or second-degree relatives with LS-associated tumors, regardless of age |
25.2.1 Natural History of Lynch Syndrome
Lynch syndrome is the most common type of hereditary CRC and is estimated, depending on the study, to represent 1–6 % of all CRC cases. It is inherited in an autosomal dominant fashion. It is highly penetrant with approximately 50 % of mutation carriers developing CRC and approximately 80 % developing CRC or a malignancy associated with Lynch syndrome [14].
The clinical features of Lynch syndrome include CRC in multiple family members in several generations. The mean age of cancer diagnosis is 45 years. Approximately 70 % of colon cancers are proximal to the splenic flexure, though it should not be overlooked that up to 30 % of Lynch syndrome-associated tumors are found in the distal colon. The histologic features of these tumors include tumor-infiltrating lymphocytes, mucinous and signet-ring features, and medullary growth patterns. In addition, progression of colonic adenomas to carcinomas is accelerated compared to the rate in sporadic CRC. Compared to an 8–10 year progression interval, in Lynch syndrome, adenomas become carcinomas in 2–3 years [15].
Although CRC is the predominant tumor, extracolonic tumors also can develop. Lynch-associated tumors are listed in Table 25.3. Endometrial cancer is the second most frequent malignancy in Lynch syndrome. Additional tumors include ovary, stomach, small bowel, pancreas, hepatobiliary, brain, and urothelial tract [16]. A subset of patients with MMR gene mutations develop cutaneous lesions, a syndrome known as Muir-Torre (see Sect. 25.6). Given the number and diversity of related tumors, clinicians need to be especially astute and query patients’ family histories carefully in order to facilitate diagnosis.
Table 25.3
Lifetime risk of extracolonic tumors reported in families with a mismatch repair mutation
Endometrial cancer | 27–71 % |
Ovarian cancer | 3–13 % |
Gastric cancer | 2–13 % |
Urinary tract cancer | 1–12 % |
Brain tumor | 1–4 % |
Biliary cancer | 2 % |
Small bowel cancer | 4–7 % |
25.2.2 Genetics of Lynch Syndrome
25.2.2.1 Inheritance
Lynch syndrome is inherited as an autosomal dominant disorder. Carriers inherit one mutated MMR gene from one parent and one un-mutated gene from the other parent. The heterozygous cell has sufficient MMR function to carry out normal DNA repair and protect the cell from excessive mutation. However, when, in a somatic precursor cell, the un-mutated gene is lost by normal mutagenic processes, which inevitably happens in the many cell divisions that occur during human development and adult life, then this cell loses all MMR function and excessive mutation ensues in the subsequent generations of this cell’s lineage. A striking hypermutability develops and increases the probability that transforming mutations will accumulate in oncogenes or tumor suppressor genes. The second hit that leads to loss of the un-mutated MMR gene results from random mutational processes, including large gene deletion, chromosome loss and re-duplication, homologous recombination proximal on the chromosome to the mutated MMR gene followed by segregation of the two chromosomes carrying the same mutated allele, nucleotide substitution, or small insertion or deletion. There are rare reports of children with biallelic MLH1 and MSH2 mutations who are prone to develop cancers at a young age. These cancers include leukemia, lymphoma, brain tumors, and gastrointestinal malignancies. There are also café au lait spots reminiscent of neurofibromatosis. This syndrome is referred to as the Childhood Cancer Syndrome (CCS) [17, 18].
25.2.2.2 MSH2, MLH1, and MSH6
In 1993, MSH2 was identified as the first Lynch syndrome-associated gene. Linkage analysis performed in two large Lynch syndrome families mapped a susceptibility locus to chromosome 2p16. Subsequent work identified the MSH2 gene in this region as a candidate locus [19]. Germline mutations of MSH2 were subsequently identified in Lynch syndrome patients. MSH2 mutations constitute the largest percentage of mutations found in Lynch syndrome. Linkage methods were also utilized to identify the MLH1 gene in Scandinavian Lynch syndrome families [20]. The gene was localized to chromosome 3p21 and subsequent work revealed germline MLH1 mutations in Lynch syndrome patients. Mutations in MSH2 and MLH1 account for the majority (over 85 %) of MMR gene mutations in patients with Lynch syndrome. In 1997, disease-causing mutations were identified in Lynch syndrome families in the MSH6 gene, which is localized within 500 kb of MSH2 on chromosome 2p21 [21]. MSH6 mutations account for approximately 10 % of all mutations in Lynch syndrome.
A multi-national database maintained by the International Collaborative Group on HNPCC (ICG-HNPCC now called InSight) was established to catalog Lynch syndrome-associated mutations. The group’s website (http://www.insight-group.org/) provides regular updates on mutations reported worldwide in MMR genes. To date, there are over 500 reported mutations primarily involving MLH1, MSH2, and MSH6. Most of the mutations cause loss of MSH2 function. The majority of these null mutations result in premature translation-termination of the protein through large multi-exon deletions or small insertions or deletions that cause reading frame shifts, nucleotide substitutions that cause stop codons or aberrations in RNA splicing. In addition, a significant proportion of the putatively disease-causing mutations cause amino acid substitutions that produce a mutant protein that has no catalytic activity. These missense mutations can pose a serious challenge in the clinical interpretation of the mutation because (1) biochemical information of the amino acid change is usually unavailable, (2) it is difficult to distinguish rare disease-causing variants from rare benign variants, and (3), if the affected is in a small family, segregation analysis lacks power.
The most common recurrent mutation in MSH2 is an A → T transversion at c.942 + 3 in the donor splice site of intron 5. The substitution disrupts normal splicing so that the mutant gene produces an abnormal mature mRNA in which exon 4 is joined to exon 6. The abnormal mRNA encodes a functionally inactive, mutant protein with an in-frame deletion of amino acids 265–314. MSH2c.942 + 3 is a hot spot for mutation. The adenine at this site is the first in a run of 26 adenines, which presumably causes the hypermutability. However, the mechanism is not understood. It is estimated that this mutation accounts for 5–10 % of all Lynch syndrome mutations and it has arisen independently multiple times on different chromosomal backgrounds. MSH2c.942 + 3A > T is the most common mutation in Newfoundland, and it occurs on one and the same chromosomal background, indicating that in this population the mutation is inherited identical by descent from a founder individual who introduced the mutation into the population many generations ago. Numerous other MMR gene mutations are known to be shared by unrelated individuals due to founder mutation in an ancestral individual, as the mutation is found on the same chromosomal background [22].
25.2.2.3 PMS2
The PMS2 gene is localized to chromosome 7p22, originally identified based on its homology with bacterial and yeast MMR genes [23]. Early work on PMS2 was complicated by the existence of a family of PMS2 related genes and a partial PMS2 pseudogene, confounding investigators who thereby missed disease-causing variants in PMS2. Mutations in PMS2 appear to predispose to Turcot syndrome and early-onset CRC (see Sect. 25.6). However, this finding is controversial as a large European group could not replicate the findings [24]. Similar to the situation for MLH1 and MSH2, biallelic PMS2 mutations cause a childhood cancer syndrome [25].
25.2.2.4 EPCAM
In 2009, two groups studied cancers that were deficient for MSH2 and MSH6 protein by immunohistochemistry or that exhibited MSI but in which no germline mutation had been identified in MSH2, MSH6, or MLH1 [26, 27]. Diagnostic multiplex ligation-dependent probe amplification (MLPA) analysis (a method that allows detection of large genomic deletions) showed a decreased signal for exon 9 of the EPCAM gene (also referred to as TACSTD1), which is the last exon in the gene that is located 16 kb upstream of MSH2, but the deletion did not extend into the MSH2 gene. Further analysis of Dutch and Chinese families showed that deletions of the 3′ end of EPCAM, which includes the polyadenylation-signal, abolish transcription termination. Failure to appropriately terminate EPCAM transcription , in turn, leads to transcription read-through into the MSH2 promoter across its CpG-rich promoter region. Inappropriate transcription of the MSH2 promoter results in induction of methylation of the MSH2 promoter and transcriptional silencing of MSH2 in cells expressing EPCAM. This unusual mechanism explains both the MSH2 methylation mosaicism (cells that express EPCAM exhibit the methylation, whereas cells that do not express EPCAM do not exhibit methylation) and the allele-specific methylation of the MSH2 promoter (MSH2 methylation occurs in cis on the chromosome that carries the MSH2 deletion and not on the intact chromosome). Moreover, this mechanism also explains why the abnormal pattern of mosaic somatic methylation segregated with the mutation in families.
A subsequent clinical study of 194 carriers of 3′ EPCAM polyadenylation-signal deletions was noted to have similar ages of colorectal cancer onset as MLH1 and MSH2 carriers [28]. In contrast, only three women with EPCAM deletions were found to have endometrial cancer leading to a lower cumulative lifetime risk compared to other MMR gene mutation carriers. EPCAM is expressed at lower levels in uterine tissue. Consequently, MSH2 silencing as result of EPCAM polyadenylation-signal deletion may occur less frequently in the uterus. Higher rates of duodenal and pancreatic cancers were noted in EPCAM polyadenylation-signal deletion carriers compared to other Lynch syndrome families. Whether screening recommendations should be different for EPCAM polyadenylation-signal deletion carriers remains to be determined. Molecular genetic testing for EPCAM deletions is recommended for the subset of families in whom germline mutations in MMR genes are suspected but have not been identified.
25.2.2.5 Genotype-Phenotype Correlations
Clinically, Lynch syndrome cancer phenotypes vary as a function of the mutated MMR gene involved. Mutations in MLH1 have been associated with higher incidence of CRC, whereas MSH2 mutations have been associated with higher rates of extracolonic tumors. MSH6 mutations have been associated with lower rates of CRC with higher rates of endometrial cancer [29], while the phenotypic pattern associated with PMS2 mutations is still unclear. Furthermore, while the mean age of colorectal cancer diagnosis in MSH2 and MLH1 mutation carriers is 43–46 years, in MSH6 mutation carriers, the mean age of cancer diagnosis is 10 years older [29]. EPCAM deletion carriers appear to have lower rates of endometrial cancer but possibly higher rates of duodenal and pancreatic tumors [28].
The reasons for these gene-specific clinical phenotypes are poorly understood and may be due to different functions of MMR proteins, modifier genes and/or environmental risk factors. Data on modifier genes in Lynch syndrome are conflicting. Candidate genes such as TP53 [30], NAT2 [31–33], and CCND1 (the gene that encodes cyclin D1) [34, 35] have been tested for modifier effects but the data are not conclusive. Environmental risk factors have also not been extensively examined, though a multicenter American study found tobacco use to be associated with higher risk of CRC development [36]. More recent data has suggested that higher body mass indices (BMIs) greatly increase the risk of colorectal adenomas, a finding that was noted only in men [37]. Further studies with large, collaborative study design are needed to elucidate the role of modifier genes and environmental factors in Lynch syndrome.
25.2.3 Management of Lynch Syndrome
25.2.3.1 Diagnosis
Diagnosis is defined by the presence of a mutation in one of the MMR genes. The diagnosis can be supported by the presence of MSI in the cancer, absence of MMR protein by immunohistochemistry (IHC), or a variety of other laboratory tests (biochemical or mRNA analysis, methylation testing, etc). Statistical models that take into account all available cancer family history and laboratory testing have been developed to estimate the probability that a person carries a mutation [38, 39] and universal testing of all incident CRC cases helps to identify mutation carriers, who are then referred to the genetics clinic. Unaffected relatives can then learn their mutation status and take preventive measures [38, 39].
There are two approaches to molecular testing for Lynch syndrome. The first is to test for a mutation in an MMR gene directly by PCR-based direct DNA sequencing and some form of analysis for large deletions (MLPA or hybridization-based assays). The sensitivity of PCR-based direct sequence analysis is 80–90 %. However, the high frequency of amino acid substitutions of unknown clinical significance reduces the specificity of this testing and additional testing (e.g., MSI testing) would be required in these cases. The other approach is to test the tumor for MSI, MMR protein staining by IHC, or both, and then perform mutational analysis on cases in which MSI is found and/or an MMR protein is absent. MSI is more sensitive than IHC because as noted above missense mutations constitute a substantial proportion of Lynch syndrome mutations. The sensitivity of MSI testing is ~90 %. Consequently, in families with compelling family history, both approaches may be used. If no germline MMR mutation is identified, a missed mutation (e.g., methylation aberration transmitted through the germline) or mutation in one of the less-often-sequenced MMR genes (e.g., PMS2) is possible. Testing of additional tumors in the family for MSI or loss of expression of MMR protein or linkage analysis can help rule out missed mutation. Since MSI or loss of expression of MMR protein can result from non-hereditary causes, namely, by somatic mutation or somatic gene silencing by methylation of the MLH1 promoter, methylation studies and/or BRAF mutation assessment can be performed to help identify such tumors [40]. Finally, advances in DNA sequencing technologies have led to the development of methods to sequence many culprit genes at one time, which provides a simplified approach to mutation identification in presumptive carriers. Indeed, it is now feasible to sequence the entire genome on a clinical basis, and the day is coming when people will have their whole genome sequenced in order to learn their genetic risks and take appropriate medical precautions.
25.2.3.2 CRC Treatment and Survival
There has been conflicting evidence about response to chemotherapy and survival in tumors with MSI (including Lynch syndrome and non-hereditary cases) compared to tumors without MSI. Survival studies do not suggest major differences in Lynch syndrome patients with CRC compared with sporadic CRC [41, 42]. In contrast, a systematic meta-analysis of MSI status and prognosis in CRC (including both Lynch syndrome-associated and non-hereditary tumors) suggested that patients with tumors that exhibit MSI had a 15 % better outcome compared with those with tumors that did not exhibit MSI [43]. As result of these studies, most oncologists withhold chemotherapy for Stage II CRCs with MSI, because the outcomes are the same if not better without treatment.
Regarding responsiveness to chemotherapeutic agents , in vitro studies have suggested that MMR deficiency reduces the cytotoxicity of 5-fluorouracil (5-FU), which is a commonly used chemotherapeutic agent for CRC [44, 45]. To date, studies on response to chemotherapy stratified by MSI status have yielded mixed results and, therefore, MSI status is not yet recommended as a predictive marker of response to chemotherapy.
Finally, immune checkpoint inhibitor therapy is now being used now for metastatic MSI-H colon cancer.
25.2.3.3 Screening
Colonoscopy remains the preferred method for screening in established or suspected Lynch mutation carriers. The current recommended screening regimen is colonoscopy every 1–2 years starting at age 20–25 years or 10 years younger than the youngest age at diagnosis in the family [46]. The recommendation for short intervals between colonoscopies is based on evidence of accelerated adenoma-to-carcinoma progression in MMR mutation carriers [47]. The relative risk reduction with colonoscopic screening was 62 % in two Finnish studies [48, 49]. This translates into a 72 % reduction in mortality attributed to colonoscopic screening with polypectomy [50, 51].
Data on screening for extracolonic cancer is less clear. Endometrial sampling for endometrial cancer, transvaginal ultrasound for endometrial and ovarian cancer, and urinalysis with cytology for urothelial malignancies are often recommended. Similarly, prophylactic colectomy, hysterectomy, or oophorectomy may be pursued but solid prospective data on the effectiveness of these surgical options is not currently available [46].
25.2.3.4 Prevention
The effect of nonsteroidal anti-inflammatory drugs (NSAIDs) as a chemopreventive agent in CRC development in MMR mutation carriers is unknown. A recent study investigating the effect of sulindac on surrogate endpoints for cancer showed increased epithelial proliferation in the proximal colon of Lynch syndrome patients taking the NSAID, which does not support a chemopreventive role for this NSAID [52]. The first large-scale, randomized control trial of aspirin and starch in MMR mutation carriers did not show differences in CRC incidence between the treatment and standard-care arms at 5 years [53]. However, subsequent follow-up data suggested a delayed benefit in aspirin users compared to placebo (Burn, unpublished data). Further studies would need to be done to elucidate the potential role of NSAIDs, aspirin, and other chemopreventive agents in Lynch syndrome.
25.3 Mismatch Repair
The MMR system recognizes and corrects incorporation errors that occur during DNA replication , some chemically induced or spontaneous DNA lesions, and mismatches produced during homologous recombination. Components of the system are also utilized during somatic hypermutation in the maturation of B-cells. MMR repairs both base-base mismatches and insertion/deletion loops (IDLs). Base-base mismatches can be generated by DNA polymerase incorporation errors that escape the proofreading activity of the DNA polymerases. IDLs occur when primer and template strands dissociate and re-anneal incorrectly. Slipped strand mispairings, as these are called, occur preferentially in repetitive DNA sequences of one-nucleotide to five-nucleotide repeats, referred to as microsatellites. Slipped strand mispairings in microsatellite regions can be of varying lengths depending on where loops form in the double helix. The high frequency of unrepaired IDLs plays an important role in development of MSI. MSI is one part of the mutator phenotype associated with MMR gene mutations, in which somatic mutations accumulate at a higher rate as a result of loss of MMR function.
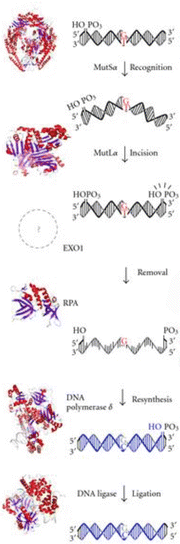
MMR pathways are conserved across all domains of life. Initially, investigation of prokaryotic MMR provided major insights into the existence and functions of genes involved in MMR. The genes mutH, mutL, and mutS were identified in E. coli, and subsequently homologues for mutL and mutS were found in yeasts, mammals, and other species. In Fig. 25.3, we depict MMR mechanisms in eukaryotes schematically. MMR involves four stages: (1) mismatch recognition, (2) incision of the strand that contains the erroneous base(s), (3) strand excision to remove the base(s), and (4) restoration of the correct base by DNA replication using the undamaged DNA template. While MMR mechanisms have been more precisely defined in prokaryotes, the focus in this chapter is on eukaryotic MMR mechanisms. Recent review articles provide in-depth discussions of MMR biochemistry in both prokaryotes and eukaryotes [54–56].