Fig. 22.1
Aldred Scott Warthin, M.D., Ph.D., (1866–1931) pioneer in hereditary cancer research.
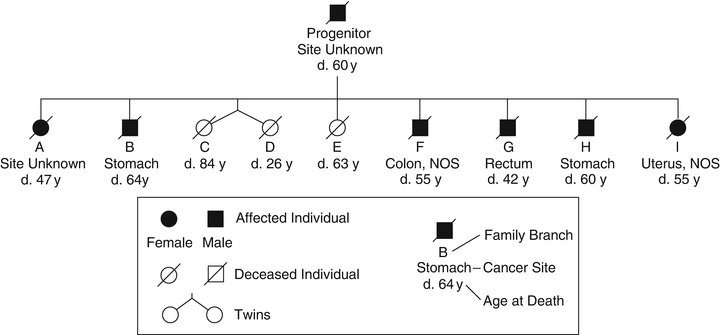
Fig. 22.2
Pedigree family G . This family was described in the first published systematic evaluation of a hereditary cancer syndrome, now known to fit criteria for Lynch syndrome. This is an abridged version of the pedigree since the updated publication contained an extremely large number of individuals. In this figure, each individual (A to I) represents a branch of the family; blackened symbol represents affected branches.
Half a century later, Lynch et al. [2] described two Midwestern families with clinical features strikingly similar to Warthin’s Family G. Individuals in these families had early-onset colorectal cancers and a litany of other cancer types, particularly gynecologic and gastric. Warthin’s successor as chairman of pathology in Ann Arbor, Michigan, invited Lynch to take possession of Warthin’s documents, which included detailed scrolls of the pedigree of Family G (Fig. 22.2), slides and cancer tissue blocks. An update of Family G was published in 1971 [3]. The family’s mismatch repair (MMR) gene mutation was finally identified in 2000 in a splice site of the MSH2 gene [4]. In 2005, a description of over a century of work on Family G was published [5]. Importantly, the pattern of cancer has continued relentlessly in Family G in accord with an autosomal dominant mode of genetic transmission.
With the continued efforts of many scientists it was finally accepted that cancer was a genetic disease. In fact, to reflect the recent revolution in cancer research and the discoveries made, it should be stated that cancer is a genomic disease, or a disease of the genome [6]. Over the last 10–15 years, breakthroughs in the identification of many important genes have significantly improved our understanding of cancer pathogenesis. At the foundation of this discovery era was the wealth of information provided by the Human Genome Project.
The intent of this chapter is to review the genes known to be associated with hereditary forms of cancer/cancer syndromes, and highlight examples of pathways in which the products of these genes interact. We also indicate where current knowledge is limited and where further investigation may be warranted.
22.2 Cancer Development
According to current models, tumor development is a continuous process of mutation accumulation that ultimately leads to cellular autonomy, unlimited growth and metastasis [7]. One of the most important events in this process is the initial destabilization of homeostasis that allows mutations to accumulate in genes that regulate cell proliferation, development, and differentiation. The genetic mechanisms involved in the earliest stages are not clear, but certain phenotypic features are notable. The cancer cell phenotype can be described by six hallmark features: self-sufficiency in growth signals, insensitivity to growth-inhibitory signals, avoidance of programmed cell death, limitless replicative potential, sustained angiogenesis, tissue invasion, and metastasis [7]. Generally it has been thought that three to seven successive mutations are required for the malignant conversion [8–10]. Various sources of spontaneous DNA damage are estimated to alter about 25,000 bases per human genome per cell per day out of the 3 × 109 base pairs in the genome [11]. However, cells are endowed with specific mechanisms for repairing the damage and for maintaining mutations at reasonable levels. Consequently, the mutational load at any given time in both germ-line and somatic cells can be viewed as a dynamic equilibrium between the extent of DNA damage and the efficiency of DNA repair [12]. In principle, the likelihood that a single cell will acquire enough independent mutations necessary for malignant cancer development is quite low and statistically unlikely, given that the normal mutation rate is approximately 1.4 × 10−10 mutations per base pair per cell generation [13]. However, it is now well recognized that this theory is contradicted by the rate of incidence of various cancers. Not surprisingly, cancer biology and genetics have found a way around these statistical predictions. It has been suggested that two major mechanisms could result in higher mutation rates that would make cancer development more probable. First, highly selective mutations could occur that enhance cell proliferation, creating an expanded target cell population for the next mutation [14]. Second, mutations could occur that affect the stability of the entire genome at the DNA and/or the chromosomal level, thereby increasing the overall mutation rate [13, 15].
Tumorigenesis is associated with alterations (mutations) in two main classes of genes—oncogenes and tumor-suppressor genes—each of which can drive neoplasia by increasing the tumor cell burden through stimulating cell division or inhibiting cell death or cell-cycle arrest.
Less than 10 % of patients with cancer have highly penetrant inherited (germ-line) mutations that predispose to cancer. Germ-line mutations of genes known to be responsible for inherited forms of cancer and their syndromes are summarized in Table 22.1. A mutation within this context is defined as any change in the sequence of the gene, including single base pair substitutions, large or small deletions/insertions and amplifications or translocations of chromosomal segments. In the germ line, the most common mutations are subtle (point mutations or small deletions/insertions), whereas all types of mutation can be found in tumor cells.
Table 22.1
Overview of inherited cancer syndromes
Cancer gene category | Syndrome | Gene (alias) | Gene location | OMIM ID | Major tumor types |
|---|---|---|---|---|---|
Tumor-suppressor genes (genomic stability and repair genes) | |||||
Ataxia telangiectasia | ATM | 11q22.3 | 208900 | Leukemias, non-Hodgkin lymphomas | |
Bloom syndrome | BLM | 15q26.1 | 210900 | Leukemia, lymphoma | |
Hereditary breast and ovarian cancer | BRCA1, BRCA2 | 17q21, 13q12.3 | 113705, 600185 | Carcinomas of breast and ovary | |
Fanconi anemia | FANCA, C or G in 90 % cases | 16q24.3, 9q22.32, 9p13.3 | 607139, 613899, 602956 | Acute myeloid leukemia, solid cancers | |
Lynch syndromes (HNPCC, Lynch I, Lynch II, Lynch III, Turcot and Muir–Torre syndromes) | MLH1, MSH2, MSH6, PMS2 | 3p21.3, 2p22-p21, 2p16, 7p22 | 120436, 120435, 600678, 600259, 609309, 609310, 614350, 614337 | Colon adenocarcinoma, uterus endometrial adenocarcinoma; skin sebaceous tumors (Muir-Torre); biallelic inheritance results in pediatric lymphomas and brain tumors (Lynch III) | |
Attenuated polyposis (MAP) | MUTYH | 1p34.3-p32.1 | 608456 | Colon adenomas and adenocarcinomas | |
Nijmegen breakage syndrome | NBS1 | 8q21 | 251260; 613065, 609135 | Lymphoma, leukemia | |
Rothmund–Thomson syndrome | RECQL4 | 8q24.3 | 268400 | Bone osteosarcoma | |
Werner syndrome | WRN | 8p12-p11.2 | 277700 | Meningiomas, soft tissue sarcomas | |
Xeroderma pigmentosum | XPA, XPC, ERCC2, ERCC3, ERCC4, ERCC5, DDB2, POLH | 9q22.3, 3p25, 19q13.2-q13.3, 2q21, 16p13.3-p13.13, 13q33, 11p12-p11, 6p21.1 | 278700, 278720, 126340, 133510, 133520, 133530, 600811, 603968 | Non-melanoma skin cancer (basal cell and squamous cell carcinomas) and cutaneous melanoma | |
Tumor-suppressor genes (other) | |||||
Familial adenomatous polyposis (FAP), desmoid disease, Turcot syndrome | APC | 5q21-q22 | 175100; 135290 | Adenomas and adenocarcinomas of colon, small intestine (and CNS for Turcot syndrome) | |
Oligodontia–colorectal cancer syndrome | AXIN2 | 17q23-q24 | 604025;608615 | Colon adenomas and adenocarcinomas | |
Juvenile polyposis | BMPR1A, SMAD4 | 10q22.3, 18q21.2 | 174900; 601299, 600993 | Juvenile polyps in gastrointestinal tract | |
Familial gastric carcinoma | CDH1 (E-cadherin) | 16q22.1 | 192090; 137215 | Diffuse gastric, and lobular breast carcinoma | |
Familial atypical multiple mole melanoma | CDKN2A (P16/INK4A, p14/ARF) | 9p21.3 | 600160; 155601 | Cutaneous melanoma, pancreatic adenocarcinoma | |
Familial cylindromatosis (Brooke–Spiegler syndrome) | CYLD | 16q12.1 | 605041; 605018 | Cylindromas of skin | |
Hereditary multiple exostoses | EXT1, EXT2, EXT3 | 8q24.11-q24.13, 11p12-p11, 19p | 133700; 608177; 133701; 608210; 600209 | Osteochondromas | |
Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) | FH | 1q43 | 136850; 150800 | Leiomyomas of skin and uterus; renal carcinoma | |
Birt–Hogg–Dube syndrome | FLCN (BHD) | 17p11.2 | 135150; 607273 | Diverse renal tumors, skin fibrofolliculoma | |
Simpson–Golabi–Behmel syndrome | GPC3 | Xq26 | 300037; 312870 | Wilms tumor, hepatoblastoma | |
Hyperparathyroidism jaw-tumor syndrome | HRPT2 (CDC73) | 1q31.2 | 145001; 607393 | Parathyroid adenoma and carcinoma, ossifying fibroma of the jaw | |
Multiple endocrine neoplasia type 1 | MEN1 (menin) | 11q13.1 | 131100; 613733 | Parathryoid and pituitary adenomas, pancreatic islet cell tumors | |
Neurofibromatosis type 1, Neurofibromatosis–Noonan syndrome, Watson syndrome, Juvenile myelomonocytic leukemia (JMML) | NF1 (neurofibromin) | 17q11.2 | 162200; 613113; 601321;162210; 193520; 607785 | Neurofibroma | |
Neurofibromatosis type 2 | NF2 | 22q12.2 | 101000; 607379 | Bilateral vestibular schwannomas; meningiomas | |
Basal cell nevus syndrome (Gorlin syndrome) | PTCH1, PTCH2, SUFU | 9q22.3, 1p34.1, 10q24.32 | 109400; 601309; 603673; 607035 | Basal cell carcinomas of skin | |
Cowden disease (CD)/Bannayan–Riley–Ruvalcaba syndrome (BRRS) | PTEN | 10q23.31 | 153480; 158350; 601728 | Breast, thyroid and endometrial carcinomas; skin trichilemmoma GI hamartomas | |
Carney complex type I | PRKAR1A | 17q24.2 | 160980; 188830 | Myxomas of the skin, heart and breast | |
Hereditary retinoblastoma | RB1 | 13q14.1-q14.2 | 180200; 614041 | Retinoblastoma (usually involving both eyes) | |
Familial paraganglioma–pheochromocytoma syndromes caused by SDHB, SDHC, and SDHD mutations | SDHB, SDHC, SDHD | 1p35-36.1, 1q21-23, 11q23 | 606864; 115310; 171300; 185470; 602413; 602690; 168000 | Multiple bilateral paragangliomas of the neck | |
Peutz-Jeghers syndrome | STK11 (LKB1) | 19p13.3 | 175200; 602216 | Stomach and intestinal hamartomatous polyps and adenocarcinomas | |
Medulloblastoma predisposition | SUFU | 10q24.32 | 607035; 155255 | Medulloblastoma | |
Li–Fraumeni syndrome, Hereditary Adrenocortical Carcinoma | TP53 (p53) | 17p13.1 | 191170; 151623; 202300 | Breast cancer, bone and soft tissue sarcomas | |
Tuberous sclerosis, lymphangioleiomyomatosis (LAM) | TSC1 (hamartin), TSC2 (tuberin) | 9q34.13, 16p13.3 | 191092; 605284; 606690 | CNS cortical hamartomas, skin subungual keratomas, renal angiomyolipomas | |
von Hippel–Lindau syndrome | VHL | 3p25.3 | 608537; 193300 | Renal clear cell carcinoma, CNS capillary hemangioblastoma | |
WAGR, Denys–Drasch, Frasier syndrome, and non-syndromic hereditary Wilms’ tumor | WT1 | 11p13 | 607102; 194070; 194080; 136680 | Wilms’ tumor (nephroblastoma) | |
Oncogenes | |||||
Familial malignant melanoma | CDK4 | 12q14.1 | 123829; 609048 | Cutaneous melanoma | |
Familial gastrointestinal stromal tumors | KIT | 4q12 | 164920; 606764; 273300 | Multiple gastrointestinal stromal tumors (GIST) | |
Hereditary papillary renal cell carcinoma (HPRCC) | MET | 7q31 | 164860; 605074 | Multiple bilateral type I papillary renal cell carcinoma | |
Familial gastrointestinal stromal tumors | PDGFRA | 4q12 | 173490; 606764 | Multiple gastrointestinal stromal tumors (GIST) | |
Multiple endocrine neoplasia type II (A and B) | RET | 10q11.2 | 164761; 171400; 162300; 142623; 188550 | Medullary thyroid carcinoma, parathyroid hyperplasia, adrenal pheochromocytoma | |
Neoplasia is initiated by a mutation in a cancer gene that results in a clonal expansion, with subsequent mutations in additional genes resulting in further rounds of expansion as part of the process of tumor progression [16, 17]. An individual with a germ-line mutation in a cancer predisposition gene has an early start on tumor development, because a mutation that can contribute to cancer is already present in every cell. As such, these individuals may readily develop multiple tumors that are detectable at an early age. In many dominantly inherited cancer syndromes, the acquired first somatic mutation affects the normal copy of the gene inherited from the unaffected parent [18].
Under normal conditions, proto-oncogenes (the normal, non-mutated form of an oncogene) stimulate cell proliferation and differentiation. These genes mainly encode proteins involved in signal transduction pathways and include growth factors, growth factor receptors, signal transducers, and transcription factors (e.g., the RET and MET genes). When mutated (activated) these genes become oncogenes and gain the ability to dominantly promote neoplastic processes. In contrast, tumor suppressors are genes whose loss of function promotes malignancy [19, 20]. They are usually negative regulators of growth or other functions that may affect invasive and metastatic potential [21]. Tumor suppressor genes can be divided into two broad functional classes: (1) gatekeepers which directly regulate cell proliferation and are rate limiting for tumorigenesis; (2) caretakers which maintain the integrity of the genome and promote cancer growth indirectly by causing an increased mutation rate [22]. Interestingly, the most common forms of hereditary cancer predisposition are caused by inherited mutations of caretaker genes.
The contribution of tumor suppressors to tumorigenesis was initially proposed by Alfred Knudson in 1971, based upon epidemiological studies of retinoblastoma [23]. According to Knudson’s two-hit model, both copies of a tumor suppressor gene need to be inactivated for tumor formation. In the familial form of a disorder associated with a tumor suppressor gene, one mutated allele is inherited from one of the parents and the second allele is somatically inactivated. When the second hit involves a large deletion or mitotic recombination (reduces the mutation to homozygosity), it is referred to as loss of heterozygosity (LOH). Knudson’s model has later been applied to other forms of familial cancer as well [18, 24]. However, not all loss-of-function mutations in tumor suppressor genes are truly recessive [25]. Evidence suggests that haploinsufficiency (functional loss of one allele) of some genes might provide a growth advantage [26]. Thus, lower gene-dosage, rather than biallelic inactivation of a tumor suppressor gene may be sufficient to exert a cellular phenotype that leads to tumorigenesis. This may be particularly true for caretaker genes, where haploinsufficiency could result in mildly increased genomic instability [27].
In contrast to classical monogenic Mendelian disorders , such as Duchenne muscular dystrophy or cystic fibrosis, wherein mutations in a single gene cause disease, a single gene defect or alteration does not cause cancer. The vast majority of genetic alterations in cancer are somatic and found only in tumor cells. In an individual with a dominant cancer predisposition syndrome, all cells in the body are a priori heterozygous, carrying one mutated and one wild-type allele [19]. The germ-line mutation can be considered as the first hit required for tumorigenesis. The next genetic change in a cell often involves the loss of the remaining wild-type allele (second step in the Knudson’s two hit hypothesis) [18]. Even though additional somatic changes are still needed, the presence of a constitutional mutation greatly quickens the process of malignant conversion, and individuals with such mutations are at higher risk of developing cancer. Mutations with strong effects are likely to cause early onset of the disease and development of multiple primary tumors; they are inherited in simple Mendelian fashion, tend to cause familial clustering of disease and show dominant pattern of inheritance [28, 29]. However, the cancers develop only after additional somatic genetic mutations occur. Importantly, clinicians must also consider the pattern of these multiple primary cancers (e.g., breast and ovarian cancers in carriers of BRCA1 and BRCA2 mutations; medullary thyroid carcinoma and pheochromocytoma in the carriers of a RET mutation, etc.) which could then guide them toward the correct diagnosis.
Most cancer susceptibility syndromes result from inherited mutations in tumor suppressor genes rather than proto-oncogenes [19]. Germ-line mutations in only a limited number of proto-oncogenes (e.g., RET, MET, KIT, PDGFRA, and CDK4) (Table 22.1), have been shown to be involved in hereditary cancer predisposition [30–32]. Although they are rare, these syndromes are of vast biological importance. The study of genes responsible for such disorders, and the cellular pathways disrupted by the mutated proteins, provides significant insights into the molecular origin and pathogenesis of both inherited and sporadic forms of cancer [28].
A subclass of tumor suppressor genes, known as caretakers or stability genes, promotes tumorigenesis in a completely different way when mutated. This group includes the mismatch repair (MMR), nucleotide-excision repair (NER), base-excision repair (BER), and homologous recombination repair genes responsible for correcting subtle errors made during normal DNA replication or induced by mutagenic exposure. In normal cells, genetic alterations are kept to a manageable minimum by stability genes, and mutation rates are much higher when these genes are inactivated [33]. As a result, all genes are potentially at risk for mutations, but only mutations in oncogenes and tumor suppressor genes will provide a selective advantage to the cancer cell.
From this, it is best to picture mutated cancer genes as contributors to cancers, rather than the sole cause. In most cases involving genes associated with hereditary cancer syndromes, the individual with the mutation faces a high probability of developing cancer, but not 100 % certainty. Penetrance is defined as the probability that a person who has the genotype will manifest the expected phenotype. For example, a carrier of a mutation in the BRCA1 gene (breast and ovarian cancer susceptibility) has an approximate 70–85 % chance of developing breast cancer during their lifetime. Although this is a very high risk, the penetrance is incomplete and not everyone with a mutation will develop the disease. There are some important exceptions to incomplete penetrance; virtually all carriers of a pathogenic mutation in the TP53 gene (Li–Fraumeni syndrome) or the APC gene (familial adenomatous polyposis coli) develop cancer. Differences in penetrance between carriers of a germ-line mutation can be associated with environmental exposures and an individual’s genetic makeup (modifier genes).
22.3 Oncogenes
Oncogenes are altered (mutated) in ways that cause the gene product to be constitutively activated or active under conditions in which the normal gene (proto-oncogene) is not active. Activation of an oncogene can result from chromosomal translocation (BCR-ABL), gene amplification (MYC), or intragenic mutations (RET) affecting key functional moieties that regulate the activity of the gene product. Activation of an oncogene through the alteration of a single allele is usually sufficient to bestow a cell with a selective growth advantage. Examples of oncogenes associated with inherited cancer syndromes and the associated tumors are included in Table 22.1. Such genes include RET and MET , which encode receptor tyrosine kinases involved in essential processes such as cell cycle progression, proliferation, differentiation, motility, and survival [34]. Many oncogenic tyrosine kinases are constitutively activated due to specific gain-of-function mutations [34, 35].
22.3.1 RET Oncogene and Multiple Endocrine Neoplasia Type 2 (MEN 2)
One oncogene that has been studied extensively within the context of hereditary cancers is the REarranged during Transfection (RET) gene , which encodes a transmembrane receptor tyrosine kinase (RTK) [36, 37]. Normal RET is expressed mainly in developing and adult neural ectoderm. RET mutations are oncogenic owing to an intrinsic gain-of-function in the receptor tyrosine kinase activity. Germ-line mutations in RET are known to cause multiple endocrine neoplasia type II (MEN2) , which consists of three distinguishable clinical phenotypes (MEN 2A, MEN 2B, and familial medullary thyroid carcinoma FMTC) [38–42]. MEN 2A is characterized by specific endocrine neoplasms (medullary thyroid carcinoma, pheochromocytoma and parathyroid adenoma); MEN 2B is characterized by neuromas of the lips and tongue, marfanoid habitus, medullary thyroid carcinoma, and pheochromocytoma; FMTC is characterized by medullary thyroid carcinoma in the absence of other tumors.
Almost all of the germ-line RET mutations that cause MEN2 are missense, but a small fraction represent small deletions or insertions that preserve the open reading frame. The mutations are concentrated in a fraction of the open reading frame. Most mutations occur in the cysteine-rich portion of the extracellular domain. Specific missense mutations with a highly focused distribution are characteristic of mutations that cause oncogenic gain-of-function, in this case in Ret RTK activity [43–45]. This contrasts with the RET mutations that cause familial Hirschsprung disease; such mutations cause loss of protein function and have a much broader distribution across the gene [46, 47]. Hirschsprung disease represents the main genetic cause of functional intestinal obstruction with an incidence of 1/5,000 live births. This developmental disorder is a neurocristopathy and is characterized by the absence of the enteric ganglia along a variable length of the intestine [48, 49]. Occasional MEN2 families with specific mutations express Hirschsprung disease.
22.3.2 RET Activation and Signaling Pathways
One of the two RET mutations commonly associated with MEN 2B results in the substitution of threonine for methionine at codon 918 (M918T) in the tyrosine kinase domain [50–52]. RET M918T, which also occurs somatically in some sporadic medullary thyroid carcinomas (MTC), leads to a Ret receptor that autophosphorylates in the absence of ligand stimulation [53]. The constitutive activation of the variant Ret RTK results in aberrant cell signaling through a series of cytoplasmic kinases, including JUN kinase, p38 mitogen activated protein kinase, Ras/extracellular signal-regulated kinase, phosphatidylinositol 3-kinase, and Akt [54].
The mechanisms of Ret signaling in tumorigenesis are known in considerable detail, since it is part of a well-studied family of RTKs. The Ret protein is a subunit in a plasma-membrane signaling complex that includes four ligands and four co-receptors [55]. The Ret intracellular domain has at least 12 autophosphorylation sites, and phosphotyrosine 1062 in particular is a binding site for many different docking proteins and is important for the oncogenic activity of Ret [56–59].
22.3.3 Genotype:Phenotype Correlations: RET:MEN 2
Activating mutations in RET can be inherited or somatic. Germ-line point mutations are causal for the dominantly inherited disorders MEN 2A, MEN 2B and FMTC [50, 51]. Mutations of cysteine residues in the extracellular domain of RET are frequently associated with MEN 2A (MTC, pheochromocytoma, and parathyroid hyperplasia). MEN 2B (MTC, pheochromocytoma, ganglioneuromas, and marfanoid habitus) is associated almost exclusively with mutations of codons 918 or 883 in the intracellular domain of RET. Genetic testing to identify patients at risk for MEN2 and FMTC has led to clinical screening and prophylactic thyroidectomy in mutation carriers [45, 60, 61].
More than ten somatic rearrangements of RET have been identified in papillary thyroid carcinomas [61]. These oncogenic mutations activate the Ret tyrosine kinase domain via different mechanisms, making Ret an excellent candidate for the design of molecular targeted therapy. Recently, various therapeutic approaches, such as tyrosine kinase inhibition, gene therapy with dominant negative RET mutants, monoclonal antibodies against oncogene products, and nuclease-resistant aptamers that recognize and inhibit Ret have been developed [62]. Two small molecule kinase inhibitors, vandetanib and cabozantinib, are approved for the treatment of locally advanced and metastatic medullary thyroid carcinoma [59].
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


