both pro- and antithrombotic effects are recognized.17 As liver disease progresses, conventional indices of the clotting cascade worsen and have been incorporated into liver disease prognostic scores including the classical Child-Pugh and the MELD scores (Table 126.1). The latter is now used to guide organ allocation in liver transplant candidates.18 The relationship between abnormal coagulation indices and progressive liver fibrosis and cirrhosis is robust across various forms of chronic liver disease.
Table 126.1 Prognostic scoring systems in cirrhosis | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
inconsistent improvement in platelet counts after transjugular intrahepatic portosystemic shunting or surgical decompression procedures.31 Reduction of splenic tissue, by splenectomy32 or partial splenic embolization,33 results in higher platelet counts, albeit with potential morbidity. Shortened platelet survival due to immune-mediated platelet destruction may be a factor in patients with severe thrombocytopenia out of proportion to the stage of hepatic dysfunction, and in some cases, there have been responses to steroids or other immunosuppression therapies.34, 35 However, there is no value in routinely measuring platelet-associated immunoglobin G in thrombocytopenic patients with chronic liver disease due to a lack of specificity of the assay. Causes for decreased platelet production include suppression of megakaryopoiesis due to decreased hepatic production of TPO, or viral illness (HCV, HIV), alcohol, or medication (alpha interferon) toxicity.36 Observations of platelet kinetics following orthotopic liver transplantation (OLT) indicate that TPO biology is disrupted in thrombocytopenic patients with advanced liver disease. A day after OLT, TPO concentrations increase followed by increased reticulated platelets and
resolution of thrombocytopenia.37 In addition, pharmacologic interventions with recombinant TPO, TPO mimetics, and IL-11 increase platelet production.28 However, TPO concentration in patients with chronic liver disease correlates poorly with platelet count,28 partly explained by analytical problems for measuring TPO.38 Therefore, it is of little value to measure TPO concentrations in thrombocytopenic patients with liver disease.
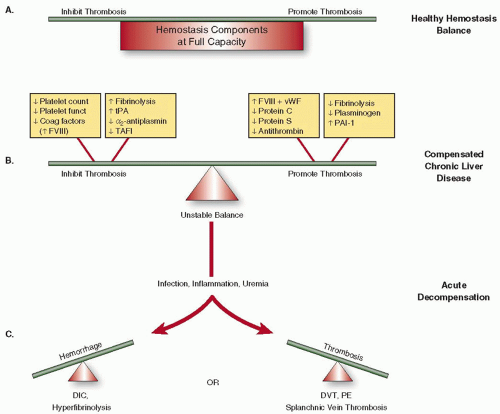 FIGURE 126.1 Changes in hemostasis balance with progressive liver disease. In healthy individuals (1A), cellular and protein components of prohemostasis and antihemostasis pathways have adequate capacities to adapt to acute physiologic stresses. In patients with compensated liver disease (1B), reductions in most coagulation/fibrinolysis factors and platelet number/function are offset by reductions in anticoagulant/antifibrinolysis factors and increased FVIII and vWF, resulting in a tenuous rebalancing of hemostasis. In patients with decompensated liver disease (1C), comorbidities further deplete most pro- and antihemostasis functions producing a fragile and imbalanced state and increased risk of hemorrhage and thrombosis. |
 FIGURE 126.2 Gastric varices with platelet plug (“nipple”) sign indicated by arrow. A scant amount of blood can be seen to be oozing from the base of the plug. (Image courtesy of Dr. Andrew Wang.) |
Table 126.2 Typical changes in activation and regulation proteins of fibrinolysis in patients with advanced hepatic dysfunction and cirrhosis | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Inherited Thrombocytopenias
Inherited Thrombocytopenias
 Unusual Sites of Arterial Occlusion
Unusual Sites of Arterial Occlusion
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


