(1)
Department of Surgery, Dar A lAlafia Medical Company, Qatif, Saudi Arabia
22.1 Introduction
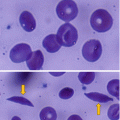
Sickle cell anemia is an inherited hemoglobinopathy that is known to be associated with long-term complications and reduced life expectancy.
The treatment of patients with sickle cell anemia generally aims to relieve their pain and manage or prevent sickle cell-related complications.
In the recent years, the management of patients with sickle cell anemia has improved markedly. This is reflected in prolonged survival of these patients, and it is attributed to several factors including:
Improvement in conservative management policies of these patients
The use of hydroxyurea
The availability of extended phenotyping of transfused donor red cells to reduce alloimmunization risks
Erythrocytapheresis to reduce iron overload in chronically transfused patients
The availability of oral chelating agents
In spite of this, patients with sickle cell anemia continue to suffer from sickle cell-related complications including the ongoing sickle cell vasculopathy.
Sickle cell anemia progression, organ damage, and sudden death can occur in these patients despite adequate supportive care measures.
Add to this the cost and availability of medical resources for lifelong medical care of these patients.
Hydroxyurea does ameliorate the disease and has been shown to reduce painful crises, acute chest syndrome, and the need for blood transfusions, but it does not provide cure.
In 1982, at St. Jude hospital, there was a patient who had leukemia and sickle cell disease. This patient had transplant using bone marrow donated from the patient’s brother. The patient was cured of both leukemia and sickle cell disease.
Hematopoietic stem cell transplantation (HSCT) is currently the only established curative treatment modality for SCA.
The benefits of HSCT are now well established, but it is associated with complications including:
HSCT-related organ toxicities
Graft-versus-host disease (GVHD)
Graft rejection (GR)
Donor availability
HSCT can establish donor-derived erythropoiesis and, more importantly, can stabilize or restore function in affected organs such as the central nervous system (CNS) and lung and prevent further deterioration of their functions.
For over two decades, hematopoietic stem cell transplantation was offered only to patients with severe SCA and only if a matched sibling donor was available.
Though human leukocyte antigen (HLA)-matched sibling donor (MSD) transplants offer the best outcomes for SCA, less than 14 % of patients with SCA have such available donors.
Currently, there are three main types of stem cell donors:
Matched related:
A brother or sister who has the same bone marrow type and the same mother and father.
Brothers and sisters are matched through special blood tests called HLA typing.
Matched Unrelated:
Volunteers who have the same bone marrow type as the patient.
Usually these types of donations are matched through national organizations that match donors and patients who have the same type of bone marrow.
Haploidentical:
Half-matched family members (usually a mother or father).
This type of donation is still considered experimental.
Stem cells can be obtained from the donor’s bone marrow or peripheral blood.
Or in some cases, stem cells are collected from the umbilical cord at the time of birth.
There are however several factors that makes bone marrow transplant not readily available option. These include:
Bone marrow transplant is not available in every country and in specialized centers within the same country. This limits accessibility.
It is a very expensive method of treatment.
Patients with sickle cell anemia are less likely to find a matching donor. This is because of the genetic complications associated with sickle cell anemia.
Regular blood transfusions which are common in patients with sickle cell anemia lead to the production of antibodies which could potentially affect the success of a future transplant.
Bone marrow transplantation is a risky procedure with a high morbidity and mortality, including the chance that about one child in ten will die from the procedure itself. The risks of bone marrow transplantation are higher in older children and adults.
One option for many patients who cannot find a suitable bone marrow match is umbilical cord blood transplant, which involves the use of blood taken from the umbilical cord and placenta minutes after the birth of a baby.
A good match is less important with cord blood transplants and known to be associated with lower risk of graft-versus-host disease or rejection.
Although bone marrow transplant is the only known cure for sickle cell anemia, the complications from a traditional bone marrow transplant can be life threatening.
Traditionally, bone marrow transplantation requires heavy doses of chemotherapy prior to transplant in order to destroy the recipient’s bone marrow so it will not reject the donated marrow. As a result of this, transplant recipients become vulnerable to life-threatening complications.
22.2 History of Bone Marrow Transplantation
Georges Mathé, a French oncologist, performed the first European bone marrow transplant in 1959 on five Yugoslavian nuclear workers whose own bone marrow had been damaged by irradiation caused by an accident at the Vinča Nuclear Institute, but all of these transplants were rejected.
Mathé later pioneered the use of bone marrow transplants in the treatment of leukemia.
Stem cell transplantation was pioneered using bone-marrow-derived stem cells by a team at the Fred Hutchinson Cancer Research Center from the 1950s through the 1970s led by E. Donnall Thomas, whose work was later recognized with a Nobel Prize in physiology or medicine.
Thomas’ work showed that bone marrow cells infused intravenously could repopulate the bone marrow and produce new blood cells. His work also reduced the likelihood of developing a life-threatening complication called graft-versus-host disease.
The first physician to perform a successful human bone marrow transplant on a disease other than cancer was Robert A. Good at the University of Minnesota in 1968.
In 1975, John Kersey, M.D., of the University of Minnesota, performed the first successful bone marrow transplant to cure lymphoma. His patient, a 16-year-old boy, is today the longest-living lymphoma transplant survivor.
The first successful hematopoietic cell transplantations (HCT) in beta-thalassemia major were reported in 1982. Since that time, more than 3000 of such transplants have been performed worldwide.
Prof. Guido Lucarelli in the 1980s was the one who popularized bone marrow transplantation (BMT) from compatible donor as the main cure for thalassemia.
Dr. Pietro Sodani used bone marrow transplantation (BMT) from haploidentical mother to child using protocol 30 to cure thalassemia.
The first patient with SCA to be treated with HCT was an 8-year-old girl who had both acute myeloid leukemia (AML) and recurrent vaso-occlusive crises. After HCT, in addition to having a normal bone marrow with no evidence of AML, her hemoglobin S level decreased to the level of the donor, who had sickle cell trait, and she had no further vaso-occlusive crises. This was published in the New England Journal of Medicine in 1984 by Johnson et al.
Additional studies with larger numbers of patients were reported in 1994 by Vermylen C and Cornu G from Belgium, with an overall and disease-free survival of 96 and 89 %, respectively.
Similar results were reported in a French study involving 14 patients by Bernaudin et al. in 1993.
Dr. Lakshmanan Krishnamurti:
Dr. Lakshmanan Krishnamurti is clinical director of hematology in the Division of Hematology/Oncology at Children’s Hospital of Pittsburgh of UPMC.
His research has focused on sickle cell disease.
Dr. Krishnamurti and his colleagues reported that six of seven sickle cell patients who received RIC bone marrow transplants in the last decade now have donor marrow and are free from symptoms of their sickle cell disease.
Dr. Lakshmanan Krishnamurti in 1999 performed the first successful bone marrow transplant on a patient with sickle cell disease at Children’s Hospital, using a rare approach known as reduced-intensity transplant.
“Bone marrow transplant is the only known cure for sickle cell disease. But doctors have avoided performing them in these patients because complications from a traditional bone marrow transplant can be life-threatening,” said Dr. Krishnamurti, director of the Sickle Cell Program at Children’s Hospital. “Through the reduced-intensity approach we developed the potential for complications is dramatically lessened”.
The first multicenter trial from the United States, published in 1996, reported results in 22 patients younger than 16 years of age who received a HLA-matched sibling HCT.
Patient selection was based on strict criteria, including patients with severe debilitating clinical events such as stroke, recurrent acute chest syndrome, and recurrent painful vaso-occlusive crises.
The conditioning regimen avoided total body irradiation and included cyclophosphamide, busulfan, and T cell depletion using either antithymocyte globulin or Campath antibodies.
A combination of methotrexate and cyclosporin or cyclosporin and prednisone was used for prophylaxis against graft-versus-host disease (GVHD).
An increased incidence of neurologic complications, with seizures in some patients, occurred; these complications were mostly resolved after prophylaxis with anticonvulsant medication.
Of the 22 patients reported, 20 survived; 16 had stable engraftment of donor hematopoietic cells, and 1 had stable mixed chimerism.
Four patients (18 %) had graft rejection, similar to the 13 % rejection rate reported among children with beta-thalassemia major who received HLA-matched sibling HCT.
Two patients died of central nervous system hemorrhage or GVHD.
The overall survival and event-free survival at 4 years were 91 and 73 %, respectively.
22.3 Selection Criteria and Indications for Hematopoietic Stem Cell Transplantation
Sickle cell anemia is a disease with an unpredictable clinical course.
This makes it difficult to choose candidates who are at significant risk for disease-related complications before they exhibit them and will benefit from bone marrow transplantation.
Taking in consideration the significant morbidity and mortality associated with bone marrow transplantation, it should be offered only to those who suffered significant sickle cell-related complications but without an end organ damage.
Hematopoietic stem cell transplantation was initially offered only to patients with severe SCA and only if a matched sibling donor was available. This is to insure better success of transplant and taking in consideration the morbidity and mortality associated with bone marrow transplant.
In the absence of a matched sibling donor, donor options include:
A matched (MUD) or mismatched unrelated donor (mMUD)Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree