Hematologic Complications
Dennis A. Casciato
Mary C. Territo
INCREASED BLOOD CELL COUNTS
I. ERYTHROCYTOSIS (POLYCYTHEMIA).
Erythrocytosis is defined as an elevation of the hematocrit and red blood cell (RBC) count above the upper limits of normal. Normal limits in adults are as follows:
|
A. Relative erythrocytosis is characterized by normal RBC mass and decreased plasma volume. Causes of relative erythrocytosis include dehydration, diuretics, burns, capillary leak, decreased oncotic pressure (“third spacing”), hypertension, and stress (“Gaisböck syndrome”). The majority of patients with Gaisböck syndrome have an RBC mass that is at the upper limits of normal and a plasma volume that is toward the lower limits.
B. Primary erythrocytosis is caused by intrinsic defects of erythroid progenitors.
1. Acquired primary erythrocytosis (polycythemia vera [PV]) is a clonal disorder. The majority of patients with PV have a somatic mutation in a gene on chromosome 9p (the JAK2 gene). Erythrocytosis develops independently of serum erythropoietin (EPO) concentration. Uncontrolled proliferation of marrow elements results in an increased RBC mass. PV, including diagnostic criteria, is discussed in Chapter 24, “Polycythemia Vera.”
2. Primary familial and congenital polycythemias result from germ line rather than somatic mutations.
a. Chuvash polycythemia is the most common congenital polycythemia in the world and the only known endemic polycythemia. The condition is named after the Chuvash population in the mid-Volga River region of Russia. The condition results from an abnormality in the oxygen-sensing pathway. Inheritance is as an autosomal recessive involving mutation of the von Hippel-Lindau (VHL) gene. Marked erythrocytosis occurs in the presence of normal to increased levels of EPO.
b. Primary erythrocytosis due to mutations of the EPO receptor (usually truncation) can be congenital or familial. Inheritance is as an autosomal dominant. An increased proliferation of erythrocytes, elevated RBC mass, hypersensitivity of erythroid progenitors to EPO, low serum EPO levels, and normal hemoglobin oxygen dissociation characterize this rare disorder.
C. Secondary erythrocytosis is associated with increased RBC mass due to extrinsic stimulation of progenitors by circulating substances such as EPO.
1. Appropriate erythrocytosis
a. Chronic hypoxemia is a potent stimulus for EPO production. Causes of hypoxemia include pulmonary diseases, right-to-left intracardiac shunts, low atmospheric0 pressure (high altitudes), alveolar hypoventilation (brain disease or pickwickian syndrome), and portal hypertension. Intermittent arterial desaturation and erythrocytosis may be caused by sleep apnea or by supine posture, particularly in obese patients with pulmonary disease.
b. Heavy smoking. Excessive and sustained exposure to carbon monoxide from cigarettes or cigars, which produces an increased affinity between the remaining oxygen and the hemoglobin molecule, is a common cause of erythrocytosis.
c. Congenital disorders include hemoglobinopathies with high oxygen affinity (abnormal oxyhemoglobin dissociation), overproduction of EPO, and familial deficiency of 2,3-biphosphoglycerate (rare).
d. Androgen therapy stimulates erythropoiesis.
e. Cobalt chloride induces tissue hypoxia and consequent EPO production.
f. Chuvash polycythemia has elements of both primary and secondary erythrocytosis.
2. Inappropriate erythrocytosis occurs with elevated EPO levels in the absence of generalized tissue hypoxia and is seen in a variety of diseases.
a. Renal diseases account for about 60% of all cases of inappropriate erythrocytosis, and renal adenocarcinomas account for half of those cases. Cysts, other tumors, hydronephrosis, and transplantation make up the remaining renal causes of erythrocytosis.
(1) Renal cell carcinomas synthesize EPO in association with erythrocytosis in 1% to 5% of cases.
(2) Renal transplantation is associated with erythrocytosis in 10% of patients. The erythrocytosis has been ascribed to transplanted artery stenosis, graft rejection, hypertension, hydronephrosis, diuretic use, and EPO overproduction from residual renal tissue, especially in polycystic disease.
b. Hepatocellular carcinoma and cerebellar hemangioblastoma each account for 10% to 20% of the cases of inappropriate erythrocytosis in the literature.
c. Other causes of inappropriate erythrocytosis are rare. Huge uterine leiomyomas and ovarian carcinoma can cause renal hypoxia or ectopic EPO production. Pheochromocytomas and aldosteronomas cause erythrocytosis through multiple mechanisms.
D. Evaluation of patients with erythrocytosis
1. Initial evaluation. The following studies are obtained in all patients with persistent erythrocytosis:
a. Perform a complete history and physical examination to search for known causes of elevated hematocrits. Search for treatments that are associated with absolute or relative erythrocytosis (androgen therapy, diuretics) and for splenomegaly, which would suggest PV. If intravascular volume depletion is suspected, replete the volume and then reassess.
b. Analyze the hemogram. The presence of granulocytosis, eosinophilia, basophilia, or thrombocytosis suggests PV.
c. Measure arterial oxygen saturation. The RBC mass is roughly proportional to the degree of arterial desaturation. Arterial oxygen saturation <90% and a PaO2 <60 to 65 mm Hg may result in erythrocytosis.
d. If the patient smokes tobacco, measure the carboxyhemoglobin concentration; values >5% are associated with erythrocytosis. Smoking may also cause granulocytosis.
e. Serum EPO level is decreased in PV and abnormalities of the EPO receptor. The concentration is normal or increased in the other disorders associated with erythrocytosis.
2. Special diagnostic studies
a. RBC mass determination previously was paramount for distinguishing absolute erythrocytosis from relative erythrocytosis. RBC mass is measured with 51Cr-labeled erythrocytes, and plasma volume is measured concomitantly with 125I-labeled albumin to assess for intravascular volume reduction. However, this useful test is becoming increasingly unavailable.
b. Abdominal radiography (ultrasonography or CT scanning) is indicated in all patients with absolute erythrocytosis that is not explained by either PV or hypoxemia because the frequency of renal causes is high.
c. JAK2 gene mutation should be sought if PV is suspected.
d. Oxyhemoglobin dissociation curve is indicated in patients with a family history of unexplained erythrocytosis.
e. Other diagnostic studies for inappropriate erythrocytosis are obtained only if the screening evaluation exposes abnormalities that could indicate pathology of a specific organ.
f. Bone marrow examination is not diagnostic of any disorder associated with erythrocytosis.
II. GRANULOCYTOSIS
A. Definitions
1. Granulocytosis. The upper limit of normal for neutrophils is 8,000/mL.
2. Leukemoid reactions. The term leukemoid reaction should be restricted to granulocytosis with circulating promyelocytes and myeloblasts.
3. Leukoerythroblastic reactions are characterized by immature granulocytes in association with nucleated erythrocytes in the peripheral blood. Platelet counts may be normal, increased, or decreased. Differential diagnosis includes the following:
a. Metastatic tumor in the marrow
b. Marrow fibrosis with extramedullary hematopoiesis
c. Marrow recovery after severe hematosuppression
d. Shock, hemorrhage
e. Brisk hemolysis, hereditary anemias
B. Causes of granulocytosis
1. Increased proliferation in the marrow is seen in myeloproliferative disorders (MPDs), in marrow rebound after suppression by drug or virus, and as a chronic response to infection, inflammation, or tumor. The mechanism of tumor-induced granulocytosis most often involves increased production of granulocyte and granulocyte-macrophage colony-stimulating factors (G-CSF, GM-CSF), interleukin (IL)-1, and IL-3.
2. Increased marrow proliferation and increased granulocyte survival are seen in chronic myelogenous leukemia (CML).
3. Shift from the marrow storage pool into the circulation is seen in response to stress, endotoxin, corticosteroids, and etiocholanolone.
4. Demargination (resulting in granulocytosis involving only mature neutrophils) is seen in stress, including emotional upset, epinephrine administration, exercise, infection, hypoxia, and intoxication.
5. Decreased egress into the tissues is seen after chronic treatment with corticosteroids.
C. Differentiation of leukemoid reactions from MPDs and CML involves complete clinical evaluation, especially for the history and presence of splenomegaly. The leukocyte differential count, neutrophil alkaline phosphatase score, and cytogenetics may be helpful (see Table 24.1). Bone marrow biopsies are frequently not discriminatory.
1. Neutrophil alkaline phosphatase scores are normal or increased in MPDs and reactive granulocytosis and decreased in CML.
2. Vitamin B12. Transcobalamins I and III are synthesized by granulocytes. The total-body granulocyte mass, when increased, is reflected by increased serum levels of vitamin B12 and unsaturated B12-binding capacity. These levels are usually elevated in patients with MPD and CML and normal in patients with erythrocytosis or granulocytosis of other causes. Transcobalamin I is increased in CML, and transcobalamin III is increased in PV.
III. THROMBOCYTOSIS
A. Thrombocytosis in cancer patients. Persistent thrombocytosis may indicate cancer. Thrombocytosis in neoplastic disease may be idiopathic or the result of bleeding or bone marrow metastases. Generally, thrombocytosis associated with solid tumors is mild, but values may exceed 1,000,000/mL.
B. Common causes of transient thrombocytosis
1. Acute hemorrhage or phlebotomy
2. Acute infection
3. Recovery from myelosuppression (viruses, ethanol, cytotoxic agents)
4. After surgery (persists for about 1 week)
5. Response to therapy for folic acid or vitamin B12 deficiency
6. Certain drugs (epinephrine, vinca alkaloids, and perhaps miconazole)
C. Causes of chronic thrombocytosis
1. Iron deficiency (the most common cause of thrombocytosis)
2. MPDs
3. Neoplasms (idiopathic or bone marrow metastases)
4. Chronic inflammatory diseases
5. Hyposplenism (postsplenectomy states, hemolytic anemias, regional enteritis, sprue, and splenic atrophy from repeated infarctions)
D. Differentiation of causes of chronic thrombocytosis. After history and physical examination, helpful screening tests for the evaluation of chronic thrombocytosis include the following:
1. Peripheral blood. Megathrombocytes and fragments of megakaryocytes are rarely seen in disorders other than the MPDs and CML. A normal mean platelet volume suggests reactive thrombocytosis. The granulocyte differential is helpful for recognizing MPDs and CML. The presence of hypochromia and microcytosis supports iron deficiency.
2. Serum iron, iron-binding capacity, and serum ferritin to evaluate for iron deficiency
3. Bone marrow aspirate examination demonstrates panmyelosis in MPDs and CML. Bone marrow biopsy may detect tumor involvement. Iron staining is unreliable in patients with cancer or chronic inflammatory diseases if the results show low or absent iron stores.
IV. EOSINOPHILIA
A. Definition. The upper limit of normal in absolute cell count is 550/mL.
B. Nonneoplastic causes of eosinophilia
1. Allergies and drug hypersensitivities
2. Skin diseases (many types)
3. Infection with fungus, protozoan, or metazoan; convalescence after a febrile illness
4. Eosinophilic gastroenteritis, inflammatory bowel disease
5. Eosinophilic pulmonary syndromes (e.g., Löffler syndrome)
6. Collagen vascular diseases, especially rheumatoid arthritis, polyarteritis nodosa, and Churg-Strauss syndrome
7. Contaminated tryptophan eosinophilia-myalgia syndrome
8. Chronic active hepatitis, pernicious anemia, immunodeficiency syndromes
9. Hyposplenism (see Section III.C.5)
10. Hypereosinophilic syndrome/chronic eosinophilic leukemia
C. Eosinophilia associated with neoplasia
1. Hodgkin lymphoma (up to 20% of cases)
2. MPDs and CML (common)
3. Acute lymphocytic leukemia and lymphoma (especially T-cell types)
4. Acute monocytic leukemia with inv(16)
5. Angiolymphoid hyperplasia with eosinophilia (Kimura disease)
6. Pancreatic acinar cell carcinoma (syndrome of polyarthritis, subcutaneous panniculitis, and peripheral eosinophilia)
7. Tumors undergoing central necrosis or metastasizing to serosa
8. Malignant histiocytosis
9. Eosinophilia related to treatment: RT to the abdomen, hypersensitivity to cytotoxic agents
V. BASOPHILIA
A. Definition. The upper limit of normal is 50/mL.
B. Causes of basophilia
1. Hypersensitivity reactions
2. MPDs
3. CML
4. Mastocytosis
5. Hyposplenism (see Section III.C.5)
6. Infections: tuberculosis, influenza, hookworm
7. Endocrine: diabetes mellitus, myxedema, menses onset
8. Miscellaneous: hemolytic anemia, ulcerative colitis, carcinoma
VI. MONOCYTOSIS
A. Definition. The upper limit of normal is 500 to 800/mL.
B. Causes of monocytosis
1. Hematologic neoplasms (leukemias, lymphomas, myeloma), myelodysplastic syndromes, immune hemolytic anemias, immune thrombocytopenia, and other hematologic disorders
2. Solid tumors with and without metastases
3. Inflammatory bowel disease, sprue, and alcoholic liver disease
4. Collagen vascular disease (including rheumatoid arthritis, systemic lupus erythematosus, polyarteritis nodosa, and temporal arteritis)
5. Sarcoidosis
6. Mycobacterial infections, subacute bacterial endocarditis, syphilis, and resolution from acute infection
7. Infections with varicella-zoster virus or cytomegalovirus (CMV)
8. Hyposplenism (see Section III.C.5)
9. Factitious monocytosis may occur when blood samples are taken from fingertips affected by peripheral vascular disease.
VII. LYMPHOCYTOSIS.
The differential diagnosis of lymphocytosis is discussed in Chapter 23, Section III.D in “Chronic Lymphocytic Leukemia.”
CYTOPENIA
Decreased formed elements in the circulating blood can result from decreased or ineffective production within the bone marrow, increased destruction of cells, or sequestration in the spleen. Patients with cancer often have a combination of these abnormalities. The type and duration of cytopenia depend on several factors (Table 34.1).
I. PANCYTOPENIA BECAUSE OF BONE MARROW FAILURE
A. Metastases to the marrow
1. Occurrence. Carcinomas of the breast, prostate, and lung are the solid tumors most likely to be associated with extensive marrow metastases. Melanoma, neuroblastoma, and carcinomas of the kidney, adrenal gland, and thyroid also frequently have marrow metastases.
2. Findings. Tumor volume in the marrow does not correlate directly with the degree of hematosuppression. Marrow metastases are often found in patients without any hematologic abnormality. Patients may have bone pain, bone tenderness, radiographic evidence of cortical bone involvement, or elevated serum alkaline phosphatase levels.
a. Bone marrow paraneoplastic alterations can result in qualitative and quantitative abnormalities in hematopoiesis. In the absence of marrow metastases, changes can develop that are comparable to those seen in the primary myelodysplastic syndromes, including myelodysplasia in all cell lines, marked reactive changes, stromal modifications, and bone marrow remodeling.
b. Desmoplastic reactions to metastases can result in myelofibrosis.
c. Bone marrow biopsy is superior to aspiration (with examination of the clot specimen) for detection of metastases; both techniques are complementary.
Cytologic preparations of bone marrow aspirates must be inspected at the edges of the smears for clumps of tumor cells. Immunohistochemical staining for epithelial markers may be helpful in identifying carcinomas.
Cytologic preparations of bone marrow aspirates must be inspected at the edges of the smears for clumps of tumor cells. Immunohistochemical staining for epithelial markers may be helpful in identifying carcinomas.
Table 34.1 Hematopoiesis, Cell Kinetics, and Bone Marrow Injury | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||
d. Peripheral blood abnormalities. Nearly all patients with solid tumors and leukoerythroblastosis have demonstrable marrow metastases. Thrombocytopenia (in the absence of RT or chemotherapy) is the next best indicator. Leukocytosis, eosinophilia, monocytosis, and thrombocytosis each is associated with positive marrow biopsies in about 20% of cases.
B. Marrow fibrosis
1. Occurrence. Extensive primary marrow fibrosis is characteristic of myelofibrosis with agnogenic myeloid metaplasia and late-stage polycythemia vera. Marrow fibrosis may also be secondary to neoplastic infiltration with leukemia or metastatic carcinoma or as a distant effect of some tumors without demonstrable tumor cells in the marrow. Secondary fibrosis in the marrow may also be seen in reaction to the following:
a. Collagen vascular disorders (particularly systemic lupus erythematosus, in which the fibrosis can reverse after treatment with high-dose corticosteroids)
b. Toxic agents (benzene, radiation, cytotoxic agents)
c. Infectious agents (especially tuberculosis and syphilis)
d. Hematologic diseases (myelodysplasia, pernicious anemia, hemolytic anemia)
e. Miscellaneous disorders (osteopetrosis, mastocytosis, renal osteodystrophy, Gaucher disease, giant lymph node hyperplasia, angioimmunoblastic lymphadenopathy)
2. Findings. Splenomegaly and a leukoerythroblastic blood smear are characteristic of marrow fibrosis of any cause.
C. Marrow necrosis
1. Occurrence. When diagnosed antemortem, marrow necrosis nearly always is due to either sickle cell disease or a malignancy, particularly a hematologic neoplasm. Systemic embolization of fat and marrow frequently occurs. The median survival of patients with marrow necrosis from a malignancy is <1 month. Patients with severe weight loss may develop gelatinous transformation of the marrow with marrow hypoplasia and fat atrophy; this condition is reversible.
2. Findings. Patients have severe bone pain in the back, pelvis, or extremities (75%), fever (70%), cytopenias, and a leukoerythroblastic blood smear.
a. Serum levels of alkaline phosphatase and lactate dehydrogenase (LDH) are usually elevated.
b. Radiographs are normal.
c. Bone marrow aspiration demonstrates characteristic findings: Individual hematopoietic cells are not recognizable, and cells with indistinct margins and intensely basophilic nuclei are usually surrounded by amorphous acidophilic material.
D. Bone marrow failure secondary to treatment. Ionizing radiation and most chemotherapeutic agents cause suppression of bone marrow function. Although recovery is usual after chemotherapy, recovery after irradiation is inversely proportional to dose and volume treated and may never be complete. Indeed, after doses in excess of 3,000 cGy, the bone marrow may be replaced by fatty and fibrous tissue. The distribution of marrow in the human skeleton is shown in Figure 34.1.
The occurrence of therapy-related myelodysplasia and acute myelogenous leukemia (AML) is even more worrisome for the development of treatment strategies. To develop this complication, the patient must have been treated long enough and then live long enough to manifest this long-term toxicity.
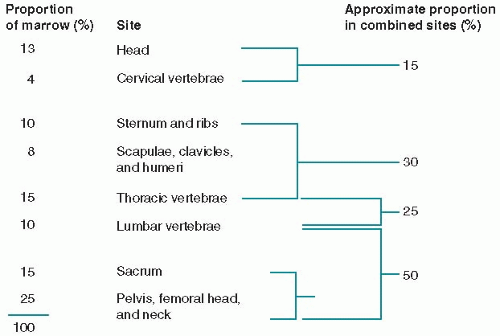 Figure 34.1. Distribution of bone marrow in healthy 40-year-old persons. Marrow cellularity is relatively decreased and amounts of fat increased in elderly subjects. (Data from Ellis RE. The distribution of active bone marrow in the adult. Phys Med Biol 1961;5:255.) |
1. Occurrence. Nearly half of the patients have a primary hematologic malignancy. The risk for AML is increased 10- to 50-fold in patients treated for multiple myeloma, Hodgkin lymphoma, non-Hodgkin lymphoma, ovarian cancer, germ cell tumors, small cell lung cancer, and childhood acute lymphoblastic leukemia (ALL). For children with ALL who achieve complete remission, the risk for therapy-related AML is greater than the risk for developing relapsed ALL.
2. Leukemogenic agents. The risk of inducing AML is directly related to the total cumulative dose and probably to dose intensity. The risk may also depend on the schedule of administration; for example, the risk in children with ALL is greatest in those undergoing weekly or biweekly therapy with epipodophyllotoxins.
a. Alkylating agents are the drugs with the most clearly demonstrated leukemogenic potential. Melphalan and chlorambucil are most often associated with AML in this class of drugs.
b. Other drugs. Epipodophyllotoxins (etoposide, teniposide), nitrosoureas, and procarbazine are also leukemogenic. Cisplatin is not a classic alkylating agent and is possibly leukemogenic; however, it is nearly always given in combination with other drugs, some of which are leukemogenic. Hydroxyurea has been implicated as being possibly leukemogenic in the treatment of MPDs, but the risk has been found to be low.
c. RT is associated with a minimally increased risk for AML when given alone but with a synergistically increased risk when combined with leukemogenic drugs.
3. Chromosome abnormalities, particularly involving chromosomes 5q or 7q, are found in 70% of therapy-linked AML associated with alkylating agents.
These same aberrations are seen in patients developing AML after exposure to leukemogenic solvents and pesticides. In contrast, certain balanced translocations involving 11q23 appear to be characteristic of myelodysplasia and AML occurring after treatment with cytostatic agents acting on DNA topoisomerase II, such as etoposide. 4. Natural history. AML usually develops 3 to 5 years after initiation of therapy but can arise after 10 years or longer; the syndrome rarely develops within 1 year. Therapy-induced AML is usually preceded by months to years of myelodysplasia. After AML develops, the course is rapid and usually refractory to treatment. Death usually occurs within 2 to 4 months of diagnosis. An important predictive factor for favorable response to intensive antileukemic therapy is the absence of a preceding myelodysplastic phase.
These same aberrations are seen in patients developing AML after exposure to leukemogenic solvents and pesticides. In contrast, certain balanced translocations involving 11q23 appear to be characteristic of myelodysplasia and AML occurring after treatment with cytostatic agents acting on DNA topoisomerase II, such as etoposide. 4. Natural history. AML usually develops 3 to 5 years after initiation of therapy but can arise after 10 years or longer; the syndrome rarely develops within 1 year. Therapy-induced AML is usually preceded by months to years of myelodysplasia. After AML develops, the course is rapid and usually refractory to treatment. Death usually occurs within 2 to 4 months of diagnosis. An important predictive factor for favorable response to intensive antileukemic therapy is the absence of a preceding myelodysplastic phase.
II. PANCYTOPENIA BECAUSE OF HYPERSPLENISM
A. Pathogenesis. Splenic enlargement from any cause (including carcinomatous metastases) may result in phagocytosis of the circulating blood cells and the development of cytopenias. Hypersplenism with severity sufficient to beg the question of splenectomy develops most often in lymphoproliferative disorders and myelofibrosis.
B. Diagnosis. The diagnosis of hypersplenism is based on clinical judgment. The only true diagnostic test for hypersplenism is improvement in the cytopenias after splenectomy.
C. Treatment
1. Indications for splenectomy for hypersplenism are all of the following:
a. The patient has palpable splenomegaly.
b. The cytopenia is severe (e.g., anemia requiring frequent transfusions; severe neutropenia associated with recurrent, serious bacterial infections; or thrombocytopenia with hemorrhagic manifestations).
c. Other causes of cytopenia have been ruled out (e.g., disseminated intravascular coagulation [DIC]).
d. A reasonable survival time after splenectomy is expected.
e. The patient’s general medical condition is satisfactory enough to make the operative mortality risk acceptable.
f. Surgeons experienced in performing splenectomy under adverse conditions are available.
2. Consequences of splenectomy
a. Postsplenectomy blood picture is characterized by Howell-Jolly bodies, neutrophilia, eosinophilia, basophilia, lymphocytosis, monocytosis, and thrombocytosis.
b. Postsplenectomy sepsis is a potentially fatal complication, especially in children younger than 6 years of age. The most common infecting organisms are Streptococcus pneumoniae and Haemophilus influenzae. The incidence of sepsis in patients with Hodgkin lymphoma who undergo splenectomy has been reported to be 1% to 3%. Immunization may be helpful. Febrile episodes must be treated immediately and aggressively.
III. PANCYTOPENIA DUE TO HISTIOCYTOSIS
A. Hemophagocytic histiocytosis is an acquired syndrome of exaggerated histiocytic proliferation and activation. The acquired form is frequently associated with systemic viral infection (particularly with Epstein-Barr virus [EBV]), although other microorganisms have also occasionally been implicated. These syndromes often develop on the background of another primary disease, such as autoimmunity, immunodeficiencies, or cancer (especially lymphomas). Abnormalities in genes
involved in immune response pathways (i.e., PRF1, STX11, and MUNC13-4) are evident in children with the genetic form of the disease and are being identified in patients with the acquired forms of the disease with increased frequency, so that patients who do develop the disease may have an underlying genetic predilection.
involved in immune response pathways (i.e., PRF1, STX11, and MUNC13-4) are evident in children with the genetic form of the disease and are being identified in patients with the acquired forms of the disease with increased frequency, so that patients who do develop the disease may have an underlying genetic predilection.
1. Pathogenesis. This is a systemic hyperinflammatory syndrome in which excessive stimulation of T cells and proliferation of these cells lead to disruption of immune regulation, cytokine storm, and systemic macrophage activation. This can lead to major organ failure of the liver, kidneys, or lung. The severity of the syndrome varies from mild to lethal.
2. Clinical findings include fever, severe malaise, myalgias, and often hepatosplenomegaly (which is less prevalent in adults than in children). At least two cytopenias are seen in nearly all cases.
a. Bone marrow biopsy is often hypocellular with an increase in marrow macrophages. The macrophages are vacuolated and contain ingested RBCs and erythroblasts (and perhaps other hematopoietic elements).
b. Lymph node biopsy shows normal nodal architecture with hemophagocytic histiocytes. These cells can also be seen on liver biopsies and may be evident in other effected organs.
c. Blood studies
(1) Acute phase reactants and proinflammatory cytokines are elevated.
(2) Triglycerides, ferritin, and LDH are frequently elevated. Fibrinogen and sodium are frequently decreased.
(3) Parameters indicating DIC are frequently present.
3. Treatment
a. Patients with mild to moderately severe disease may recover in weeks if the infectious agent is treatable, or the disease may resolve naturally if the patient’s immune system is intact.
b. Patients receiving immunosuppressive therapy may require drug dosage reduction.




