Research regarding head and neck squamous cell carcinoma (HNSCC) has shifted from the detection of individual gene mutations and deletions to detailing the complex interactions of networks of genes altered by genetic alterations, including sequence alterations, chromosomal aberrations, epigenetic modifications, microRNA changes, and even mitochondrial mutations. High throughput, whole-genome-based discovery approaches that have been successful in other tumor types are currently being employed in the study of HNSCC.
With improved understanding of HNSCC has come the solidification of several different mechanisms of carcinogenesis. Although the majority of these cancers still come from mutagenic environmental exposures, namely tobacco (smoking or chewing) and use of betel products, the rise in prevalence of oropharyngeal cancer in the United States is largely related to human papillomavirus (HPV) type 16. With the different etiologies of cancer development, we have seen distinctions in clinical behavior and thus can directly see how the underlying molecular mechanisms may have important clinical implications.
In this chapter, we highlight some of the newer developments in HNSCC research that have special relevance to clinical therapeutics. As we further understand the underlying genetic basis of this disease, we can transition this knowledge into developing targeted therapy and refine treatment paradigms. These data are by no means comprehensive, as the expansion in detailed knowledge regarding HNSCC biology has dramatically expanded.
GENETIC SUSCEPTIBILITY
Despite the well-established association between HNSCC and smoking, heavy ethanol use, and betel use, the overall incidence of HNSCC remains relatively low. Thus, there must be elements of genetic susceptibility that predispose certain patients to disease progression while in others there is relative resistance to cancer formation. Several case-control studies have demonstrated that first-degree relatives of patients with HNSCC were 3.5- to 3.8-fold more likely to also develop HNSCC even when controlling for factors such as age, gender, ethnicity, tobacco, and alcohol use.1,2,3 Likewise, HNSCC patients were found to be 3.8 times more likely to develop a second primary tumor if one or more first-degree relatives suffered from upper aerodigestive tract cancer.4
These familial susceptibilities provide evidence that there are likely underlying genetic mechanisms that preclude one to cancer formation. However, direct evidence of heritable HNSCC syndromes is rare. The most common direct, genetic association with HNSCC exists with Fanconi anemia, a rare, autosomal recessive disease associated with aplastic anemia, congenital anomalies, and a predisposition for cancer development, especially head and neck and anogenital squamous cell carcinoma.5 There has been suggestion that one of the mechanisms of cancer development in Fanconi anemia is HPV-related, but recent studies show that the precise mechanism of susceptibility remains unknown and that the presence of high-risk HPV viruses in Fanconi anemia-related HNSCC is variable.6 There have also been associations with HNSCC and other syndromes such as Lynch-II, Bloom, xeroderma pigmentosum, ataxia telangiectasia, and Li-Fraumeni,7 all of which have specific genetic aberrations that link them to cancer. The fact that a consistent genetic mechanism remains elusive in HNSCC highlights its heterogeneity.
Building on the link between these known syndromes and gene alterations, investigators have looked to other genetic polymorphisms, such as single nucleotide polymorphisms (SNPs), to evaluate whether patterns of HNSCC susceptibility can be detected. It is important to note that it is generally not known what the exact differential function is of these SNPs and whether they have a direct link toward carcinogenesis. Many of the single-institution studies also suffer from smaller sample sizes and lack the ability to control for the potential ethnic or geographic variability inherent in these polymorphisms.
The general families of gene alterations have focused on carcinogen-detoxifying mechanisms (cytochrome p450 members [CYP], glutathione-S-transferases [GSTs], alcohol and ethanol dehydrogenases [ADH and ALDH]), DNA damage repair, nucleotide excision repair enzymes (NER), excision repair cross-complementing rodent repair deficiency complementation genes (ERCC), x-ray repair complementing defective repair in Chinese hamster cells (XRCC), and RecA homolog, Escherichia coli (Rad51), inflammation/angiogenesis (cyclooxygenase-2 [COX-2], hypoxia-inducible factors [HIF], cytokines), apoptosis, cell cycle, and many other pathways salient to cancer formation.
Larger cooperative studies, across institutions, help to further refine the most important polymorphisms and provide the greatest evidence for their role in carcinogenesis. A cooperative European group (ARCAGE) surveyed 115 polymorphisms in 62 selected genes in a cohort of 1,511 cases and 1,457 controls.8 Several genes showed promise (CYP2, murine double-minute 2 [MDM2], and tumor necrosis factor [TNF]), but there still remains work to be done in looking at the mechanisms and applicability of these markers. Another multinational study pooled patient samples from several institutions and used a cohort of over 3,000 HNSCC patients along with over 5,000 controls and discovered a protective effect from two of the studied ADH polymorphisms.9
Despite the numerous studies looking at these relationships, the effects of polymorphic variants seem to be quite modest, and there are often conflicting studies reported on the same polymorphisms, underscoring the complexity of HNSCC as well as the role of these DNA changes. Further study to validate the previously studied SNPs with a relationship to HNSCC as well as mechanistic confirmation of an altered function would help to further our understanding of carcinogenesis. With newer technology, there will be an opportunity to perform further genome-wide screens to discover new targets as well in an effort to make sense of the complicated pathway associations that these SNPs have in the context of specific patient variables.
TABLE 14.1 COMMON GENETIC ALTERATIONS IN HEAD AND NECK SQUAMOUS CELL CARCINOMA
Alteration
Frequency
Comments
p16 inactivation
70%
Via homozygous deletion and less frequently promoter methylation
p53 mutation
50%
Predominantly mutation
High-risk HPV integration
25%
Found predominantly in oropharyngeal sites
EGFR axis alteration
80%-90%
Via amplification, overexpression, and downstream target activation
Tumorigenesis in the head and neck is the result of multiple genetic and epigenetic alterations of molecular pathways in the squamous epithelium.10,11 The progression of head and neck cancer is thought to result from multistep alterations in tumor suppressor genes and oncogenes.10,12,13,14,15,16,17,18 A variety of genetic changes have been reported for squamous cell carcinomas (SCCs) including loss of heterozygosity or amplification of specific chromosomal regions, although tumor suppressor genes have not been characterized for most of the regions that are commonly lost in HNSCC. Nevertheless, several signaling pathways have been implicated and are described in the following sections (Table 14.1).
P16/p53/Cyclin D
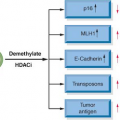
Loss of 9p21, resulting in inactivation of the p16 gene, is the most common genetic alteration in the progression of head and neck cancer.19,20 P16 is an inhibitor of cyclin-dependent kinase (CDK), which is intimately involved in G1 cell-cycle regulation. Phosphorylation and inactivation of pRb by unbridled CDK4 and CDK6 enable cells to escape senescence. Loss of chromosome 9p21 occurs in the majority of invasive tumors and is also present at a high frequency in the earliest definable lesions, including dysplasia and carcinoma in situ.19 Loss of p16 appears necessary for immortalization of keratinocytes.21 Loss of p16 protein has been observed in most advanced premalignant lesions.22 In addition to deletions and point mutations, p16 is also inactivated by methylation of the 5′CpG region.23 This methylation is associated with complete block of p16 transcription and appears to be a common mechanism for p16 inactivation. The notion that p16 inactivation is directly involved in the progression of primary tumors has been strengthened. Lack of p16 protein was detected by immunostaining in most primary invasive lesions, and tumors with absent p16 protein contained a homozygous deletion, methylation, or point mutation of p16.24
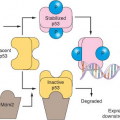
Loss of p53 on chromosomal region 17p13 and subsequent point mutation within the remaining allele is another critical step in tumor progression. Inactivation of p53 now represents the bestdescribed and most common genetic change in all of human cancer.25 Initially by analysis of exons 5-8, p53 mutations were observed in approximately 50% of head and neck tumors.26,27 The p53 gene can be inactivated by a large variety of distinct mutations and more thorough sequence analyses of exons 2-11, a p53 mutation rate of almost 80% has been observed in head and neck tumors.28p53 normally halts cell-cycle progression in the setting of DNA damage and induces apoptosis with inadequate DNA repair. p53 mutations result in a progression from preinvasive to invasive lesions, while increasing the probability of further progression. If 17p loss or p53 mutation is present in early lesions, the chance of progression to cancer within 10 years approaches 80% (33-fold relative risk). In a large definitive collaborative group study, disruptive p53 mutations were an independent prognostic marker and predicted a worse outcome in surgically resected primary tumors.29
Carcinogens in tobacco and alcohol have a causal role as the prevalence of p53 mutations is greater in patients who smoke and drink alcohol.30 HPV16- and HPV18-induced SCC of the oropharynx is more common in nonsmokers and is not associated with p53 mutations. Instead, the viral oncoprotein E6 promotes the accelerated, ubiquitin-mediated, degradation of p53.
Amplification of chromosome region 11q13 containing cyclin D1 is seen in approximately one-third of head and neck tumors.31 The role of cyclin D1 in the progression of human cancer is now well established and constitutive activation of oncogene cyclin D1 has been shown to confer a growth advantage in SCCs.32 Other tumor suppressor genes, including Rb and p16, are negative regulators of the cyclin D1 pathway and often are inactivated in human neoplasms. Cyclin D1 amplification is independent of p16 inactivation in head and neck cancers.33
PI3-K/AKT/mTOR
Mutation in the PI3-K signaling pathway are found in up to 30% of all human cancers, and activation of the PI3-K/AKT/mTOR pathway has also been implicated in tumorigenesis of HNSCC.34 Mutations of the PI3-K network have shown to confer a growth advantage, transforming capacity, and drug resistance.35 The PI3-K family is divided into three different classes based on structure and substrate specificity. Class I PI3-Ks are heterodimers of a p85 regulatory subunit and a p110 catalytic subunit, which is mutated in many cancers. The class I PI3-Ks are activated by tyrosine kinase receptors, including epidermal growth factor receptor (EGFR) and oncogenic proteins, and lead to the production of the lipid second messenger, phosphatidylinositol 3-phosphatase (PIP3), which in turn facilitates phosphorylation and subsequent activation of AKT. The PI3-K pathway also leads to activation of the serine/threonine kinase, mammalian target of rapamycin (mTOR), which in turn phosphorylates p70S6K, a kinase that modulates protein synthesis.
Invasion/Metastasis
Metastasis is a complicated, multistep process in which selective pressures select for a clone of malignant cells selected to survive in a distant, permissive environment. The multistep process includes angiogenesis, altered cellular adhesion, cellular motility, disruption of the base membrane/extracellular matrix, and anchorage-independent proliferation.
Up-regulation of hypoxia-inducible factor 1-α (HIF-1α), induced by intratumor hypoxia, has been documented in invasive HNSCC and correlates with progression to a more invasive and aggressive phenotype.36,37 HIF-1α is a master regulator of oxygen homeostasis and activates genes involved in angiogenesis, glucose metabolism, cell survival, invasion, cell renewal, and immortalization.38,39,40
The vascular endothelial growth factor (VEGF) pathway is critical in angiogenesis in HNSCC. Increased expression of VEGF and its receptors is regulated by HIF-1α-dependent and -independent pathways, both of which converge on the PI3-K/AKT pathway.
Diminished cell-to-cell adhesion, through down-regulation of cellular adhesions molecules such as E-cadherin, is integral to invasion.41,42 In addition to cell-to-cell adhesion mediated by cadherins, integrins that mediate cell-to-extracellular matrix interaction also play a fundamental role in tumor cells gaining access to the angiolymphatic system.43 Laminins, which are extracellular glycoproteins, are one of the ligands for integrins. Laminin 5 is overexpressed in invasive fronts, is associated with poorer prognosis, and downstream activates mitogen-activated protein kinase, which leads to cell survival and proliferation.44,45,46,47,48,49,50,51 In addition to altered cell adhesion, migration mediated by the Rho family of GTPases is an important step in the multistep process of metastasis.52 Cancer cells also actively disrupt the base membrane through the proteolytic activity of zinc-dependent endopeptidases, matrix metalloproteinases (MMPs), to disseminate. MMPs degrade most components of the base membrane, including collagen. Evasion from anchorage-dependent survival is a feature of metastatic HNSCC, and anoikis resistance is crucial in the process of metastasis as a defense against microenvironmental death stimuli. E-cadherin has been implicated in conferring anoikis resistance in HNSCC by physically associating with a number of signaling effectors such as PI3K and EGFR.53,54
Epidermal Growth Factor Receptor
The EGFR is one of the best-studied oncogenes in HNSCC. This receptor tyrosine kinase belongs to the ErbB family of cell surface receptors and has many downstream signaling targets associated with carcinogenesis. Once phosphorylated, the receptor can signal via the MAPK, Akt, ERK, and Jak/STAT pathways (Fig. 14.1). These pathways are related to cellular proliferation, apoptosis, invasion, angiogenesis, and metastasis.55,56,57 Expression of EGFR is a normal finding in many tissues including the dermis, gastrointestinal tract, and kidneys. However, dysfunction of this receptor and its associated pathways occurs in most epithelial cancers55 and 80% to 90% of HNSCC specifically.9,56 The story of EGFR is promising in that our understanding of its molecular biology has led directly to clinically beneficial targeted therapies and its use as a marker and prognosticator of disease.
Initially, EGFR was first found to be up-regulated in HNSCC cell lines and in a high percentage of primary HNSCC.58,59,60 Further study showed that histopathologically normal mucosa adjacent to cancer had a high degree of overexpression61 and that the up-regulation of EGFR occurs in the transition from dysplasia to HNSCC.62 Now it is well known that elevated levels of expression predict a worse disease-free and cause-specific survival.63 Studies looking at copy number amplification have also been shown to be associated with poorer prognosis in HNSCC. One study has demonstrated that the overexpression of EGFR is a biomarker for an improved response to therapy and could serve as a predictive marker to separate patients into different arms of therapeutic trials.64
In addition to serving as a marker for prognosis, EGFR axis alterations are currently under investigation as markers for response to treatment and as therapeutic targets. Several strategies exist for targeting the EGFR pathway including the use of specific tyrosine kinase inhibitors (TKIs), monoclonal antibodies blocking receptor dimerization, and antisense oligodeoxynucleotides or siRNA blocking mRNA expression.
Cetuximab is one of the most well studied monoclonal antibodies directed against EGFR. A recently published phase 3 clinical trial examined the effects of this drug in conjunction with radiotherapy in the treatment of locoregionally advanced HNSCC. This study demonstrated an overall survival benefit (49 vs. 29 months) and increased duration of locoregional control (24.4 vs. 14.9 months) in the cetuximab plus radiotherapy arm versus the arm receiving radiotherapy alone. This was the first randomized study showing a survival benefit with an EGFR targeting agent in locally advanced HNSCC.65,66 Conversely, the TKI gefitinib has shown no survival benefit for recurrent or metastatic HNSCC.66
Only gold members can continue reading. Log In or Register to continue