Gynecologic Cancers
Kunle Odunsi
Tanja Pejovic
Matthew L. Anderson
Gynecologic cancer research has mirrored all cancer research programs in that it has focused largely on molecular defects in oncogenes, tumor-suppressor genes, and DNA repair mechanisms. Several research groups have also channeled their resources into various carcinogenic phenomena such as apoptotic pathway defects, growth signaling, angiogenesis, tissue invasion, or metastasis. These efforts have led to a broad understanding of the chromosomal and molecular abnormalities that underlie malignancies of the female genital tract (vulva, vagina, cervix, uterus, ovaries, and fallopian tubes). It is clear that an improvement in outcome of these malignancies can only be achieved if (1) early diagnosis is achieved, (2) there is accurate prediction of progression and response, and (3) new treatment options reflecting the molecular pathogenesis and progression are developed. This requires detailed disease-specific understanding of the diverse molecular changes in gynecologic malignancies that ultimately lead cells to develop the following hallmarks of cancer: abnormalities in self-sufficiency of growth signals, evasion of apoptosis, insensitivity to antigrowth signals, limitless replicative potential, sustained angiogenesis, and tissue invasion and metastases. Moreover, there is growing evidence for the concept of cancer immunosurveillance and immunoediting based on (1) protection against development of spontaneous and chemically induced tumors in animal systems and (2) identification of targets for immune recognition of human cancer.1 This concept is supported by several studies in gynecologic cancers and has opened new avenues for the development of novel biomarkers and therapeutic targets. It is the purpose of this chapter to highlight and summarize some of the recent basic findings in gynecologic malignancies, with an emphasis on clinically applicable developments.
OVARIAN CANCER
Origins of Epithelial Ovarian Cancer
Epithelial ovarian cancer (EOC) arises primarily from the ovarian surface epithelium (OSE), with a subset possibly originating in the adjacent fimbria.2,3 The OSE forms a monolayer surrounding the ovary, but is composed of relatively few cuboidal cells (107 cells per ovary, or 0.05% of the entire organ). Developmentally it derives from the celomic epithelium, which also gives rise to the peritoneal mesothelium and oviductal epithelium.4 The OSE appears generally stable, uniform, and quiescent, though it can undergo proliferation in vivo.5 Despite the small number of cells within the OSE and their apparent inactivity, the risk for EOC is nearly 2%, suggesting a high malignant potential. The basis for such a high potential is poorly understood. No physiological role for the primate OSE has been established,6 and the lack of any obvious function could contribute to the asymptomatic nature of early stage EOC.
In other organs, such as colon, distinct premalignant lesions have been identified and found to accumulate genetic defects that ultimately result in malignancy. However, the search to identify similar epithelial precursors in the human ovary has proven only partially fruitful, in large part because normal ovaries are only rarely biopsied or examined. Histologic findings consistent with a preinvasive lesion for ovarian cancer have been described by a number of studies where ovaries were removed from women who eventually developed peritoneal carcinomas, ovaries from high-risk women who were undergoing prophylactic oophorectomy, and in areas of ovarian epithelium adjacent to early-stage ovarian cancers that demonstrate a transition from normal to malignant cells.7 The hypothesis that these lesions are premalignant is strengthened by observations that regions of epithelial irregularity express levels of p53 and Ki-67 intermediate between those found in normal ovarian epithelium and ovarian cancers.
Each of these observations is consistent with the hypothesis that, similar to cancers originating in other organs, ovarian cancer evolves from an intraepithelial precursor. If so, improved means to detect and/or eradicate these lesions may prove fruitful for preventing ovarian cancer.
Molecular Pathways to Ovarian Cancer
Inherited Syndromes of Ovarian Cancers
Linkage analysis of familial breast and ovarian cancers provided some of the first insights into the molecular basis of ovarian cancer. These efforts ultimately identified two gene products, BRCA1 and BRCA2, each clearly associated with an increased incidence of ovarian cancer. Although only a minority (8% to 10%) of diagnosed ovarian cancers are familial, most (76% to 92%) familial ovarian cancers are associated with mutations at the BRCA1 locus, located on 17q21. Hundreds of mutations in BRCA1 have now been identified, most commonly loss of function nonsense or frameshift mutations. Two specific mutations, 185delAG and 5382insC, are found in 1% and 0.1% of Ashkenazi Jewish women.
Functionally, BRCA1 regulates p53, an oncogene frequently implicated in ovarian cancer. Thus, loss of BRCA1 allows DNA damage to accumulate via a loss of its activation of p53. However, mutations in BRCA1 also likely contribute to ovarian cancer by mechanisms other than its interactions with p53. These include its ability to specifically regulate X chromosome gene expression mediated by an association of Xist with the inactive X chromosome.8 Consistent with this observation, sitespecific dysregulation of X-linked gene expression in BRCA1-associated epithelial ovarian malignancies have been described.
Although mutations in BRCA1 or BRCA2 are only rarely observed in sporadic, nonfamilial ovarian cancers, it is possible that the mutations in pathways by which BRCA1 regulates X-chromosome gene expression do, in fact, contribute to this disease. Characterization of genome-wide patterns of gene expression in sporadic breast cancers has allowed investigators to classify these tumors as either BRCA1-like or BRCA2-like in the patterns of their gene expression. This observation implicates the contribution of alterations in other components of the BRCA1 or BRCA2 regulated pathways to sporadic breast cancers and, possibly, ovarian cancer. Any understanding of the role of BRCA1 in ovarian cancer is further complicated by reports of women with high-risk mutations in BRCA1 who fail to develop ovarian cancer. These observations speak clearly to the role of genetic modifiers in determining whether BRCA1 or BRCA2 mutations ultimately lead to malignancy. For example, CAG repeat polymorphism in the androgen receptor has been shown to modify the subsequent risk of ovarian cancer in women with known mutations in BRCA1.
Genomic Instability
Genomic instability, manifested as a cell’s ability to tolerate DNA damage, is a hallmark of all cancer, including epithelial ovarian cancers. Tolerance to DNA damage can be achieved by alterations in any of the six major DNA repair pathways: base excision repair, mismatch repair, nucleotide excision repair, homologous recombination, nonhomologous recombination, and translesion DNA synthesis. The specific DNA pathway affected often predicts the specific type of mutations observed in particular cancers, its sensitivity to drugs, as well as clinical outcome of affected patients.
Fanconi Anemia DNA Repair Pathway
Studies on the pathogenesis of rare inherited DNA repair disorders, such as Fanconi anemia (FA), have helped define the molecular basis of defective DNA damage responses linked to cancer risk. FA is a rare genetic disorder characterized by skeletal anomalies, progressive bone marrow failure, cancer susceptibility, and cellular hypersensitivity to DNA cross-linking agents. To date, 13 FA genes have been cloned: FANCA, -B, -C, -D1, -D2, -E, -F, -G, -J, -L, -M, -N, and -I. Of these, FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, and FANCM form a nuclear core complex. Although the functional scope of this complex has not been fully defined, it is clear that it must be completely intact to facilitate monoubiquitination of the downstream FANCD2 and FANCI proteins, a change that allows FANCD2 and FANCI to colocalize with BRCA1, BRCA2 (and presumably FANCJ and FANCN), and RAD51 in damage-induced nuclear foci.9
Four lines of evidence link the FA pathway with ovarian carcinogenesis. first, BRCA2 has been identified as the FA gene FANCD1. As a result, heterozygotes for BRCA2 mutations have a high risk of tissue-specific epithelial cancers, while homozygotes develop FA. Second, an increased prevalence of epithelial cancers, including ovarian malignancies, has been observed in FANCD2 nullizygous mice. Functionally significant silencing of FANCF in ovarian cancer through promoter hypermethylation has also been described. Lastly, low levels of FANCD2 protein are found in ovarian surface epithelia from women at risk for ovarian cancer. Taken together, these data suggest that the FA pathway is important in defining predisposition to ovarian (and breast) cancer, and that aberrations of FA genes may account for some familial ovarian cancer cases not accounted for by BRCA1 and BRCA2 mutations.
In sporadic ovarian cancers, the epigenetic silencing of FA pathway through methylation of the FA gene promoter region is one of the frequent mechanisms of inactivation. One study found that 4 of 19 primary ovarian carcinomas had FANCF methylation, although a larger study of 106 ovarian tumors did not identify loss of FANCF expression. Loss of BRCA2 mRNA and protein has been reported in 13% of ovarian carcinomas, and in contrast to other FA genes, methylation is not a cause of the protein loss. Epigenetic silencing of
BRCA1 through methylation was found in 23% of advanced ovarian carcinomas (Fig. 23.1).
BRCA1 through methylation was found in 23% of advanced ovarian carcinomas (Fig. 23.1).
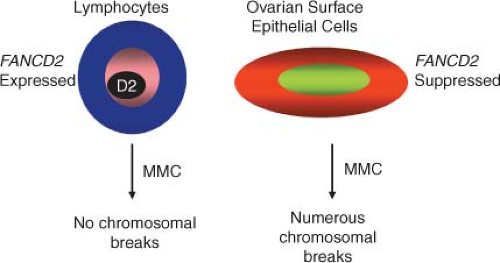 FIGURE 23.1 Tissue-restricted genetic instability in ovarian epithelial cells from women at risk for ovarian cancer with no BRCA mutations. Mitomycin C-induced chromosomal breakage is high and FANCD2 levels are low in ovarian epithelial cells but normal in peripheral blood lymphocytes and may antedate the onset of overt carcinoma. (We thank Dr. Grover Bagby for helping to create this figure.) |
Interestingly, tumors with inactivated BRCA2 are responsive to cisplatin. However, due to their low accuracy of DNA repair, these cells accumulate secondary genetic modifications that can lead to reversal of BRCA2 mutation, allowing these cells to acquire resistance to crosslinking agents.10
Other DNA Repair Pathways
Similar to the FA/BRCA pathway, disruptions of other DNA repair pathways have been observed in ovarian cancer. These disruptions account, at least in part, for the specific drug sensitivity of the tumors. Recent studies indicate translesion DNA synthesis defect in ovarian cancer is a consequence of elevation in activity of POLB, an error-prone polymerase. Inhibition of POLB in these cells results in resensitization to cisplatin.11 Overall it is believed that although inactivation of one DNA repair pathway may confer advantage to tumors, cancer cells may rely more on other repair pathways. Therefore, inactivation of the second pathway would be deleterious for these cells, causing synthetic lethality. An RNA interference screen identified the ataxia-telangiectasia mutated (ATM) pathway to be synthetically lethal with FA.12 Similarly a strategy for synthetic lethality is under investigation, using base excision repair poly(adenosine diphosphate-ribose) polymerase 1 (PARP1) inhibitors in the treatment of homologous recombination deficient ovarian cancer.13,14 PARP inhibition has been shown to be up to 1,000 times selectively more toxic to cancer cells than to wild type cells. PARP inhibitors act by exploiting a tumor cell’s defect in homologous recombination, a type of DNA repair. This is because following PARP inhibition, cells require homologous recombination to repair common types of DNA damage. Although normal cells can use homologous recombination for repair of this damage and survive, certain types of tumors (e.g., those with BRCA1 or BRCA2 defects) have lost the ability to repair by homologous recombination and will die.
Genome Wide Association Studies.
The identification of common ovarian cancer susceptibility variants may have clinical implications in the future for identifying patients at greatest risk of the disease. In this regard, several genome wide association study (GWAS) have been performed in ovarian cancer. The most striking of these was a recent study by the Ovarian Cancer Association Consortium to identify common ovarian cancer susceptibility alleles.15 A total of 507,094 single nucleotide polymorphisms (SNPs) were genotyped in 1,817 cases and 2,353 controls from the United Kingdom; and 22,790 top ranked SNPs were also genotyped in 4,274 cases and 4,809 controls of European ancestry from Europe, the United States, and Australia. Twelve SNPs were identified at 9p22 associated with disease risk (P <10−8). The most significant SNP (rs3814113; P = 2.5 × 10−17) was genotyped in a further 2,670 ovarian cancer cases and 4,668 controls, confirming its association (combined data odds ratio [OR] = 0.82 95% confidence interval [CI], 0.79 to 0.86; Ptrend = 5.1 × 10−19). The association was strongest for serous ovarian cancers (OR 0.77; 95% CI, 0.73 to 0.81; Ptrend = 4.1 × 10−21).
Transcriptional Profiling of Ovarian Cancer Histologic Subtypes
Several gene expression studies using cDNA microarrays have been performed in ovarian cancer. Additionally, several studies have focused on the alterations demonstrated in the DNA copy number.16,17,18 Array-based technology has shown that the different histological subtypes of ovarian carcinoma are distinguishable based on their overall genetic expression profiles. A common finding among several studies is the ability to distinguish low-grade serous ovarian carcinoma from high-grade carcinoma based on their gene expression profiles.17,19,20,21,22,23 A number of genes shown to be differentially expressed in EOC are known to be involved in many important cellular mechanisms, including cell cycle regulation, apoptosis, tumor invasion, and control of local immunity.20,24,25
Increased mutagenic signaling by receptor tyrosine kinases plays a major role in ovarian carcinogenesis (Table 23.1). Overexpression of
epidermal growth factor receptor (EGFR) (ERBB1), ERBB2/HER2/neu, and c-FMS has been reported repeatedly in ovarian cancer. One of the major downstream mediators of signaling initiated by these receptors is the phosphatidylinositol 3-kinase (PI3K)-AKT pathway. Aberrations in this pathway including increased AKT1 kinase activity, AKT2 and PI3K amplification, and PI3KR1 mutations may provide opportunities for therapeutic intervention.
epidermal growth factor receptor (EGFR) (ERBB1), ERBB2/HER2/neu, and c-FMS has been reported repeatedly in ovarian cancer. One of the major downstream mediators of signaling initiated by these receptors is the phosphatidylinositol 3-kinase (PI3K)-AKT pathway. Aberrations in this pathway including increased AKT1 kinase activity, AKT2 and PI3K amplification, and PI3KR1 mutations may provide opportunities for therapeutic intervention.
TABLE 23.1 GENETIC ALTERATIONS IN OVARIAN CANCER | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
It has been reported that more than 75% of ovarian carcinomas are resistant to transforming growth factor-beta (TGF-β),26 and the loss of TGF-β responsiveness may play an important role in the pathogenesis or progression of ovarian cancer. In addition, it has been shown that TGF-β1, the TGF-β receptors (TβR-II and TβR-I), and the TGF-β signaling component Smad2 are altered in ovarian cancer. Alterations in TβR-II have been identified in 25% of ovarian carcinomas, whereas mutations in TβR-I were reported in 33% of such cancers.27 Protooncogene transformation might lead either to an overexpression of mitogenic molecules or an inactivation of those with inhibitory action, thus contributing to neoplastic transformation and development. The most important protooncogenes of the first group are undoubtedly constituted by FMS and HER2/neu. The first one encodes a transmembrane tyrosine kinase receptor, which binds MCSF. It is possible that FMS-MCSF both stimulates epithelial cell proliferation and induces a chemical attraction for macrophages that, in turn, can produce mitogenic stimulating factor. Elevated plasma concentrations of MCSF are present in the sera of 70% of patients with ovarian cancer.28 The second protooncogene, HER2/neu, encodes another tyrosine kinase, which is similar to EGFR. Its action may consist of amplification of mitogenic action in target cells; this oncogene is overexpressed in 30% to 35% of ovarian cancer and is associated with a poor prognosis.29
Metastasis of Ovarian Cancers
Metastasis is the functional hallmark of all cancer. In general, metastasis involves the invasion of transformed epithelial cells across their basement membrane, through the underlying stroma, and into blood vessels and lymphatic channels, which subsequent disseminate them to distant sites. Only a tiny fraction of cells released into circulation by a tumor ever results in metastasis; understanding the mechanisms by which those cells can land and grow is a priority for cancer researchers. Given the unique need to accommodate the survival of exfoliated cells as well as their subsequent attachment and growth, it seems reasonable to assume that expression and functional organization of molecular pathways important for promoting the metastasis of ovarian cancers will differ from breast and other cancers that depend on hematogenous or lymphatic dissemination. Nonetheless, a wide variety of gene products implicated in the metastasis of other cancers have also been implicated in the metastasis of ovarian cancer. These include growth factor receptors such as EGFR,




Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
