45.1
Introduction
More than 75 years ago, osteoporosis associated with endogenous glucocorticoid excess was described by Cushing . More common is the increased fracture risk associated with exogenous glucocorticoids, a therapy used for many inflammatory disorders. Despite advances in biologically targeted therapeutics for these diseases, glucocorticoid-induced osteoporosis (GIOP) remains the most common secondary form of osteoporosis . For patients with postmenopausal osteoporosis, the fracture risk rises slowly over the years after menopause . In contrast an increased risk for fracture can be demonstrated as early as 3 months after commencing therapy with oral glucocorticoids .
As described here, the pathophysiology of GIOP is complex, but there are effective measures for prevention and treatment. Despite the heightened fracture risk and the presence of good treatments, the standard of care is low, and many patients fracture. Unlike the other side effects of glucocorticoid therapy, such as worsening of congestive heart failure or diabetes mellitus, osteoporosis causes no symptoms until there is a fracture. This chapter reviews the pathophysiology and management of this important disorder and reports on attempts to improve the standard of care.
45.2
Epidemiology of glucocorticoid-induced osteoporosis
45.2.1
Association of glucocorticoids with bone mass, bone turnover, and fractures
GIOP is the most common form of drug-induced osteoporosis . At any given time an estimated 0.2%–0.5% of the general population use oral glucocorticoids chronically (typically defined as ≥3 months) . Persons with rheumatoid arthritis (RA), polymyalgia rheumatica, temporal arteritis, systemic lupus erythematosus (SLE), and other chronic rheumatic disorders comprise more than half of chronic glucocorticoid users in the US population. Asthma, chronic obstructive pulmonary disease (COPD), inflammatory bowel disease, and a variety of inflammatory skin disorders (i.e., pemphigus vulgaris, pemphigoid, and atopic dermatitis) also constitute a large proportion of chronic, and an even greater percentage of acute, glucocorticoid use. In the United States, more than 50% of glucocorticoid prescriptions are written by generalists .
Dependent in part on glucocorticoid dose, during the first 6–12 months of therapy, there is an initial loss of 1.5%–3% of bone mineral density (BMD) . Trabecular bone is preferentially affected, followed ultimately by losses in cortical bone . The literature is divided, however, on whether trabecular bone is lost most rapidly from the trochanter or the lumbar spine . Bone loss is at least partially reversible by lowering or discontinuing the glucocorticoid . Biochemical markers of bone formation decline abruptly following short-term oral and even inhaled and intraarticular glucocorticoid administration, but they return to near baseline once the glucocorticoids are discontinued . Biochemical marker changes in GIOP do not correlate well with BMD changes , and a significantly heightened fracture risk may persist for a year or more following even shorter term glucocorticoid discontinuation . Following approximately 2 years of glucocorticoid therapy, the rate of bone loss decreases in many patients to approximately 1.5%–3% per year, dependent on residual dose. However, BMD continues to be lost at a rate higher than that of normal aging. Glucocorticoid-induced bone loss occurs in men and in premenopausal women . However, people who already have very low bone mass [e.g., postmenopausal women not on hormone replacement therapy (HRT)] are more likely to reach a fracture threshold sooner. Studies of glucocorticoid dose effects are confounded by the variable timing of glucocorticoid administration, differing disease process, variable alternative osteoporosis risk factors (independent of glucocorticoid use), cotherapies (e.g., cyclosporine), and the fact that fracture risk is determined by factors other than just BMD. As the bone field moves more toward a construct of absolute rather than relative risk (RR) prediction, scoring rules based on an amalgam of risk factors can successfully be applied to better discern fracture risk in persons using glucocorticoids as well .
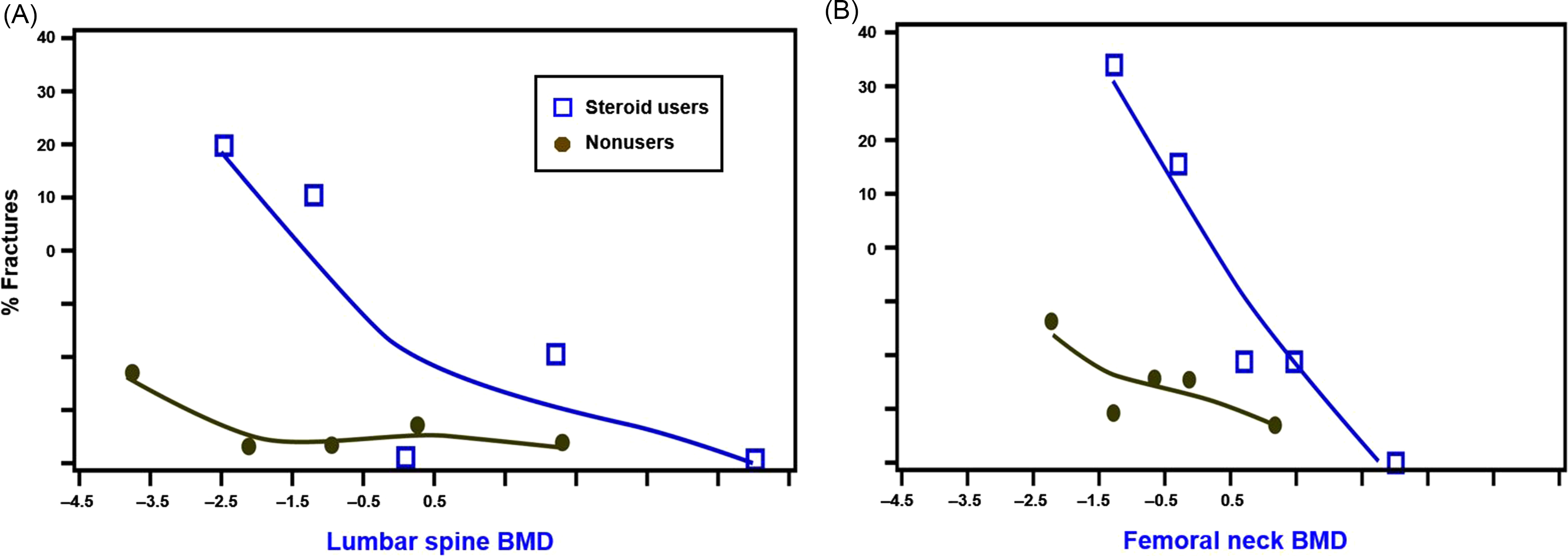
Because glucocorticoids affect both bone quantity and quality , fractures are the outcome measure of greatest importance. Observational studies suggest that more than 40% of long-term users will ultimately fracture . A meta-regression of the control arms of GIOP randomized controlled trials document a greater than 5% annual incidence of new vertebral fractures among patients newly initiating glucocorticoids and a 3% annual incidence of nonvertebral fractures in those who are chronic users . However, some studies observed a greater, 15% incidence of morphometrically defined vertebral fractures after only 1 year in patients on median doses of less than 10 mg/day . A small study of giant cell arteritis found a nearly 40% 1-year fracture rate among these mostly older adults being treated with high-dose glucocorticoids . Subjects participating in clinical trials do not represent the full spectrum of glucocorticoid users. The preponderance of evidence indicates that glucocorticoid-treated patients experience fractures at a higher BMD threshold than nonusers ( Fig. 45.1 ) . However, some studies have refuted the premise of a higher BMD fracture threshold among glucocorticoid-treated patients .
45.2.2
Effects of glucocorticoid dose and routes of administration
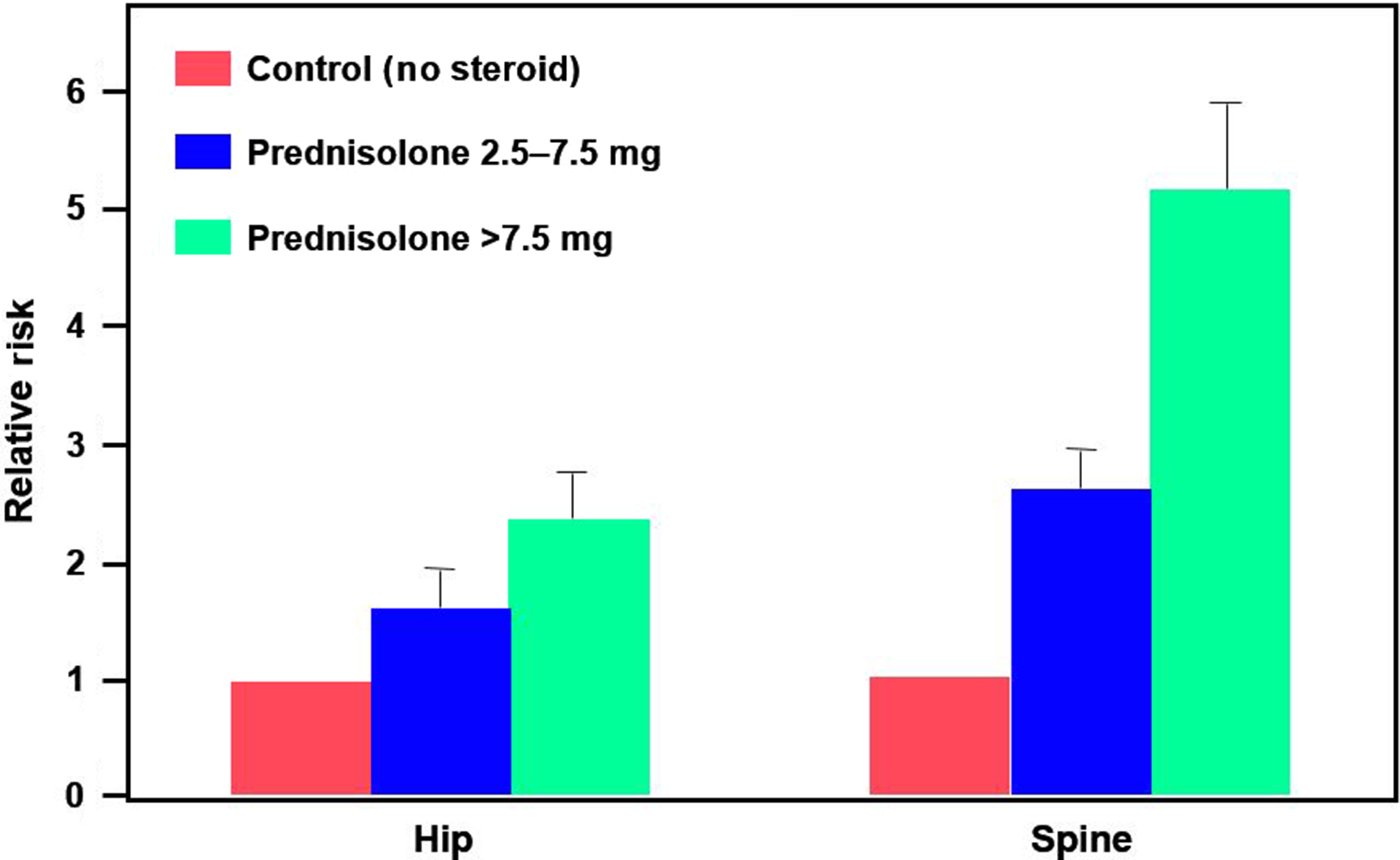
As described in the introduction, oral glucocorticoids in low doses may lead to demonstrably increased fracture risk after as few as 3 months of therapy . A study of the UK General Practice Research Database (GPRD) identified more than 240,000 glucocorticoid users who were matched by age, gender, and clinical practice to a similar-sized comparison cohort . Patients were divided into three groups: those taking <2.5 mg of prednisolone daily (low dose), those taking 2.5–7.5 mg daily (medium dose), and those taking >7.5 mg daily (high dose). Interestingly, the control group consisted of patients receiving glucocorticoids by methods thought to have minimal systemic effects: topical, otic, ophthalmic, or nasal administration. The RR of both hip and spine fractures increased in a dose-dependent fashion, beginning at 3 months, including hip fracture [RR, 1.61; 95% confidence interval (CI), 1.47–1.76] and vertebral fracture (RR, 2.60; 95% CI, 2.31–2.92). There was a trend toward increased risk even below the physiologic range of 2.5–7.5 mg/day of prednisolone per day ( Fig. 45.2 ). It should be noted that 2.5 mg of prednisolone is equivalent to approximately 3.1 mg of prednisone.
These epidemiologic data are supported by a study in which bone formation markers were measured in subjects given graded doses of prednisone. In women, 5 mg of prednisone daily was enough to depress serum levels of the bone formation markers osteocalcin, N-terminal propeptide of type I procollagen, and C-terminal propeptide of type I procollagen. Indeed, earlier studies showed that one dose of prednisone given to a normal person could affect bone formation markers. These studies provide evidence that a safe dose of glucocorticoid for bone may not exist. However, for patients with primary or secondary adrenal insufficiency, replacement doses of glucocorticoids generally are considered safe for bone.
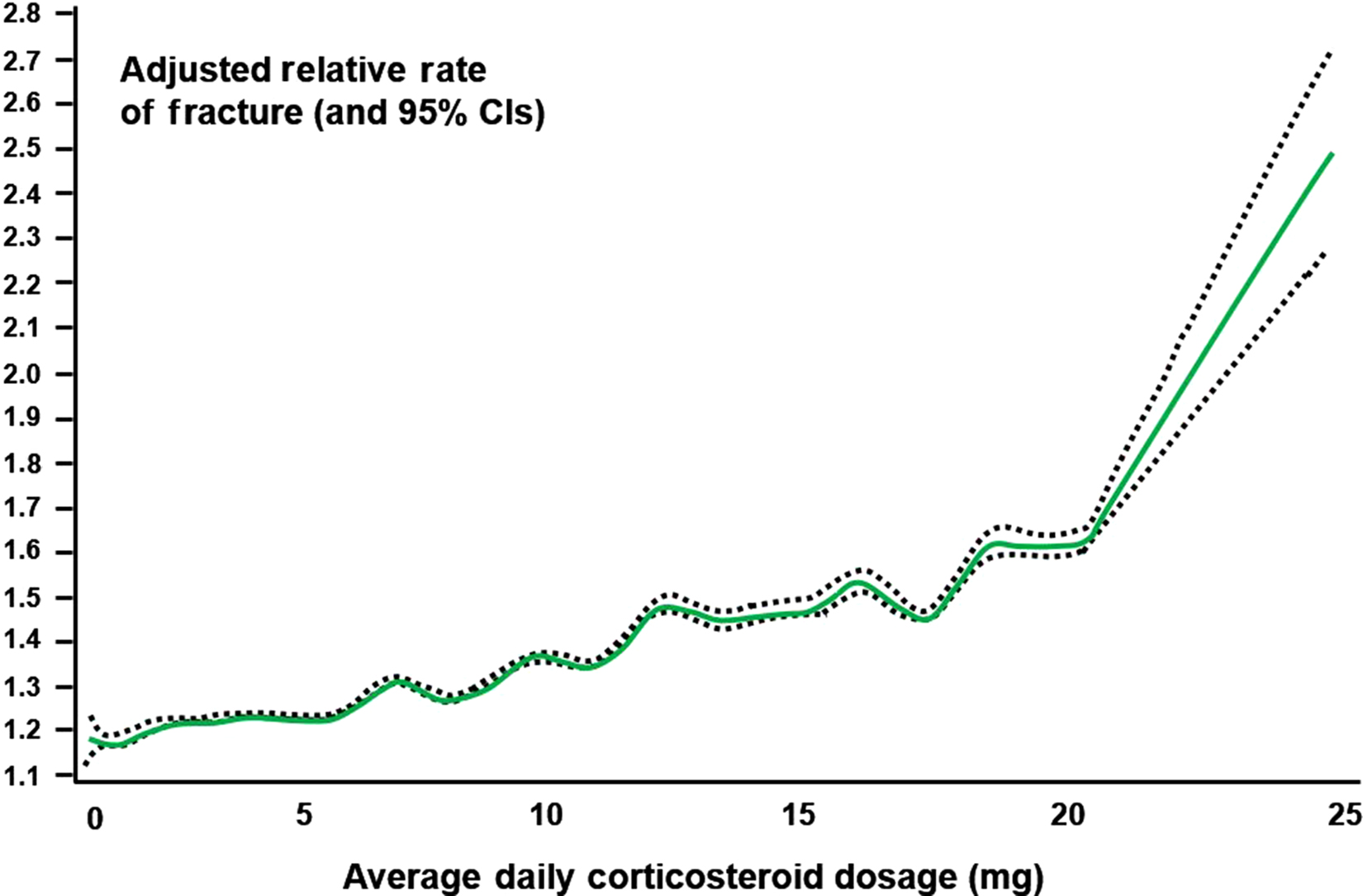
Debate continues on whether peak, current, or cumulative dose is most predictive of fracture. Cumulative glucocorticoid dose appears to be the most important predictor of bone loss based on several studies . Patients getting over 15 mg/day of prednisone equivalent, who have had at least 1 g of cumulative prednisone equivalent, have a nearly threefold increased risk of hip fractures compared to nonusers . In an analysis of nearly 18,000 people enrolled in US health plans, hip and other nonvertebral fractures occurred with glucocorticoids in both a dose- and time-dependent fashion . Alternate-day dosing does not fully spare bone . Further analyses from the UK GPRD database found that adverse effects of glucocorticoids on bone occurred rapidly and were most strongly related to daily rather than cumulative dose . In the GPRD study a monotonic relationship was seen between clinical fractures and glucocorticoid dose up to 20 mg/day; after that point the association increased in a more exponential fashion ( Fig. 45.3 ). Other observational studies confirm a 15–20 mg dose of prednisone as an inflection point in the risk of spine and hip fractures . It should be noted, as evidenced by a median glucocorticoid usage duration of approximately 30 days, that many glucocorticoid users in this study had dermatologic conditions rather than systemic inflammatory diseases . Even shorter term “pulses” of high-dose intravenous glucocorticoids or relatively short courses of oral therapy (between 130 and 500 mg of cumulative prednisolone) led to a high rate of bone loss or an elevated risk of hip fractures, respectively. The effects of inhaled, topical, and rectal steroids are discussed later. The admonition from clinical guidelines (e.g., Adler and Hochberg ) to preferentially use topical in lieu of systemic glucocorticoids is generally supported by the literature. However, high doses of inhaled glucocorticoids and, rarely, skin and mucous membrane preparations may have deleterious effects on bone, but the underlying conditions, concomitant or intermittent oral glucocorticoid courses, and improper inhaled administration technique (with more drug ingested) may increase the chances for low bone density and/or fracture.
45.2.3
Bone outcomes in diseases that commonly require glucocorticoid use
45.2.3.1
Rheumatoid arthritis
RA patients constitute the largest group of chronic glucocorticoid users. RA patients on glucocorticoids have lower BMD than those not on these agents . In a large Norwegian RA cohort, current glucocorticoid use was associated with significant loss of BMD at both the total hip [adjusted odds ratio (OR)=2.6] and spine (adjusted OR=2.7) . An increased rate of fractures has also been observed in observational studies . A large cohort study of RA patients showed that a woman taking an average dose of 8.6 mg of prednisone has a nearly 33% chance of a self-reported clinical fracture after 5 years of follow-up . At least two retrospective studies identify fractures as one of the most commonly documented adverse events of therapeutic glucocorticoid use . In a multicenter cross-sectional study of RA patients, vertebral deformities were found in 25% of patients on glucocorticoids versus in 13% of controls . The occurrence of vertebral deformities was dose dependent; every 1 mg prednisolone equivalent daily increased the adjusted OR. Among RA patients receiving glucocorticoids, the hands and forearms and other peripheral sites may be partially spared from bone loss and fracture. Of note, observational studies of fracture outcomes may be prone to selection bias and confounding by indication because “sicker” RA patients are more likely to be prescribed glucocorticoids and have an adverse (fracture) outcome.
There is particular controversy about whether a safe glucocorticoid dose exists for persons with RA . Some experts suggest that glucocorticoids may even protect bone in RA patients by improving functional status and reducing circulating proinflammatory cytokines deleterious to bone. Independent of glucocorticoids, RA causes both regional and generalized bone loss . RA patients are at higher risk for fracture and vertebral deformity . A number of observational studies , metaanalyses , and several RA clinical trials have failed to identify an adverse effect of low-dose (defined as <10 mg/day prednisone equivalent ) glucocorticoids on bones of RA patients. However, none of these clinical trials was large or long enough to fully clarify the magnitude of the fracture risk; some did not systematically assess fracture outcomes in all patients; and in at least one , patients were allowed to use bone-protective medications.
As evidenced by population-based cohort and case–control studies of general populations showing that glucocorticoids lead to an approximately twofold increased risk of fractures independent of age, gender, and the presence of RA , it is likely that RA and glucocorticoids (at modest to higher doses) are both independent and significant risk factors for osteoporosis and fractures.
45.2.3.2
Polymyalgia rheumatica and giant cell arteritis
Among the other rheumatic diseases, bone loss is also commonly noted with polymyalgia rheumatica and giant cell (temporal) arteritis . Of 120 giant cell arteritis patients followed between 1950 and 1981, fractures were detected in 38%, the most common glucocorticoid adverse event among these patients . The considerably older age and the high doses often initiated for giant cell arteritis (common starting dose of 60 mg/day of prednisone) are strongly related to a high risk of osteoporosis morbidity in this clinical population, in particular.
45.2.3.3
Systemic lupus erythematosus
Glucocorticoids are associated with significant bone loss and fractures in populations of predominately younger, and disproportionately African American, women with SLE . In one study, 12% of lupus patients followed for approximately 6000 person-years experienced fractures—a rate fivefold higher than in the general US population . For each decade of cumulative prednisone use at 10 mg/day (36.5 g), there was a 2.5-fold increase in fractures in another SLE cohort . Older age and longer glucocorticoid use (rather than peak or average dose) were stronger determinants of fracture risk. In a British lupus cohort a cross-sectional analysis confirmed a 9% prevalence of fragility fractures . Indeed, lupus patients, matched for age, have similarly reduced BMD as RA patients .
45.2.3.4
Asthma, chronic obstructive pulmonary disease, and other respiratory disorders
Persons with asthma and COPDs and, of lesser prevalence, sarcoidosis, cystic fibrosis, and other chronic respiratory conditions comprise the second largest category of chronic glucocorticoid users overall . A 12%–50% of COPD patients have prevalent vertebral fractures . Smoking, low body mass index, and diminished sunlight exposure are important contributors to this high fracture risk. Chest radiographs, obtained regularly for pulmonary manifestations, afford an additional opportunity to identify occult compression fractures in many chronic lung disease patients .
Several studies have suggested that some types of patients using inhaled glucocorticoids may have lower BMD than those who do not use such agents. Although safer for bone than oral or parenteral glucocorticoids , inhaled glucocorticoids have biological effects on bone , particularly among postmenopausal women . In a prospective study of patients with COPD, inhaled triamcinolone was associated with lower BMD in spine and hip. In at least one case–control study, inhaled corticosteroid use was associated with a significant risk for fractures (OR, 1.19; 95% CI, 1.10–1.28) . In a clinical trial of etidronate to prevent bone loss and fractures among asthma patients, there was a 17% rate of new fractures even among patients receiving only inhaled glucocorticoids. The equivalent of inhaled fluticasone of 2000 µg/day for 7 years or of triamcinolone acetonide 1200 µg/day for 20 years decreased BMD by approximately 10% in two of the more carefully conducted observational studies . In investigations of inhaled glucocorticoids, it is challenging to identify pure users of inhaled glucocorticoids because most heavy users of inhaled glucocorticoids receive periodic “bursts” of oral therapy. Fluticasone appears to have greater glucocorticoid potency and may be more toxic to bone than other preparations . However, some prospective observational studies and a metaanalysis have not confirmed an effect of inhaled glucocorticoids on BMD or fractures , although studies are limited by short durations of follow-up and confounding effects of respiratory disease activity and severity . Of note, only 5.3% of the 266 studies reviewed in the metaanalysis met quality criteria for the analysis.
45.2.3.5
Inflammatory bowel disease
Patients with inflammatory bowel disease (e.g., Crohn’s disease and ulcerative colitis) are at increased risk of osteoporosis and fracture . Despite advances in pharmacotherapy of these disorders, systemic glucocorticoids are commonly used for active disease and substantially contribute to bone loss, although other factors, such as malabsorption of calcium and/or vitamin D, play a significant role as well . Although not completely benign to bone, controlled-release budesonide may have fewer deleterious bone effects than other glucocorticoid preparations in inflammatory bowel disease . Although there have been reports of systemic absorption of rectal glucocorticoids for distal colitis, a more recent study did not demonstrate changes in bone turnover markers. Specifically, whereas both prednisolone and hydrocortisone administered rectally were able to improve colitis over a 2-week period, neither glucocorticoid affected bone formation markers (serum osteocalcin and bone-specific alkaline phosphatase) nor a bone resorption marker (urinary deoxypyridinoline).
45.2.3.6
Inflammatory skin disorders
Topical glucocorticoid preparations are considered to have very few, if any, systemic effects in adults. Nonetheless, long-term use of large amounts of glucocorticoid preparations to the skin, particularly to very thin skin areas such as the scrotum, may have rare systemic effects on other tissues, including bone. For example, topical clobetasol was reported to cause clinical Cushing’s syndrome . It is more likely that patients with skin diseases will be at risk for osteoporosis if they are treated with oral glucocorticoids .
45.2.3.7
Epidemiology of glucocorticoid-induced osteoporosis prevention and treatment
Despite data on the effectiveness of antiosteoporotic therapies in GIOP (see Section 45.6 ), only 5%–62% of even the highest risk patients on chronic glucocorticoids (typically defined as >3 months of therapy) in the United States, Canada, Europe, and Latin-America receive therapies to prevent or treat GIOP . Among those at the highest risk (e.g., women and men older than 70 years of age), only 32% received bisphosphonates or HRT in one study . This gap in internationally recommended care is seen in both community and university medical centers . Although there are many reasons for the paucity of GIOP preventive actions and for the significant practice pattern variation in GIOP management, symptomatic glucocorticoid toxicities such as mood changes, weight gain, insomnia, hypertension, and hyperglycemia often receive more attention from patients and physicians . Osteoporosis does not cause symptoms until there is a fracture. In addition, some physicians still do not recognize osteoporosis as a significant consequence of glucocorticoid use and do not properly counsel patients about this risk . Although practice pattern variations exist across specialties, certain prescribers of glucocorticoids, such as gastroenterologists and dermatologists, provide GIOP preventive management to fewer than 20% of their at-risk patients .
Efforts to improve quality of care in GIOP have met with mixed results. An intervention in Southern Tasmania, Australia, provided educational materials, GIOP guidelines, and academic detailing to both physicians and pharmacists within the region and assessed pre- and postintervention changes in calcium and vitamin D supplementation and prescription therapy . An adjacent region (Northern Tasmania) was used as the control. Substantial improvements in the use of calcium supplementation and pharmacologic therapies (31%–57%) were observed in hospitalized patients between the pre- and postintervention time periods. This study was limited by assessment of only hospitalized patients and lack of patient or provider-level randomization. Nevertheless, the results suggest that evidence-based guidelines and locally endorsed educational materials in conjunction with academic detailing to both physicians and pharmacists may be effective in improving GIOP management. Another trial randomized 21 rheumatologists caring for 373 chronic glucocorticoid users to an intervention consisting of a lecture, discussion, and confidential physician audit of practice patterns . In this academic medical center, no differences in the rates of BMD testing or prescription therapies for GIOP were observed in the intervention compared to the control group. An attempt to involve community pharmacists in improving GIOP management resulted in a significant change in calcium use but not in the use of prescription antiosteoporosis agents .
In the United States, 153 physicians (following 799 chronic glucocorticoid users) were dynamically randomized on the Internet to receive either a control intervention or a GIOP intervention consisting of tailored case-based continuing medical education, feedback on rates of past GIOP testing and treatment with achievable benchmarks , and an educational “toolbox” focused on improving prevention of GIOP. Intention-to-treat analyses showed that intervention versus control physicians had similar rates of BMD testing [19% vs 21%, P =not significant (NS)] and osteoporosis therapy use (26% vs 24%, P =NS). Among the physicians completing the three full intervention modules, intervention physicians had higher rates of BMD testing (26% vs 16%, P =.04), bisphosphonate prescribing (24% vs 17%, P =.09), or met the combined endpoint of BMD or osteoporosis therapy (54% vs 44%, P =.07) compared to control physicians. Thus this Internet-based intervention had little effect on improving quality of care for patients at risk of GIOP in the intent-to-treat population. However, physicians with more intense exposure to the intervention trended toward higher rates of GIOP management compared to control physicians . It is difficult for interventions to improve GIOP management to be cost-effective in many health-care systems . Health systems interventions using fracture liaison service health professionals to assist with GIOP management could offer future promise .
The substantial morbidity and mortality related to fractures along with good clinical evidence that treatments do work make GIOP prevention and treatment a high priority for providers caring for patients receiving glucocorticoids chronically. Novel strategies to improve suboptimal GIOP management are needed to reduce the disparities between evidence-based GIOP guidelines and actual practice. For example, there is growing interest in the use of fracture liaison services throughout many countries, and whether such a model that might identify glucocorticoid users and refer them for bone care would work at improving care in this area remains to be seen .
45.3
The pathogenesis and molecular basis of glucocorticoid action on bone metabolism and development
45.3.1
Glucocorticoid mechanisms of action
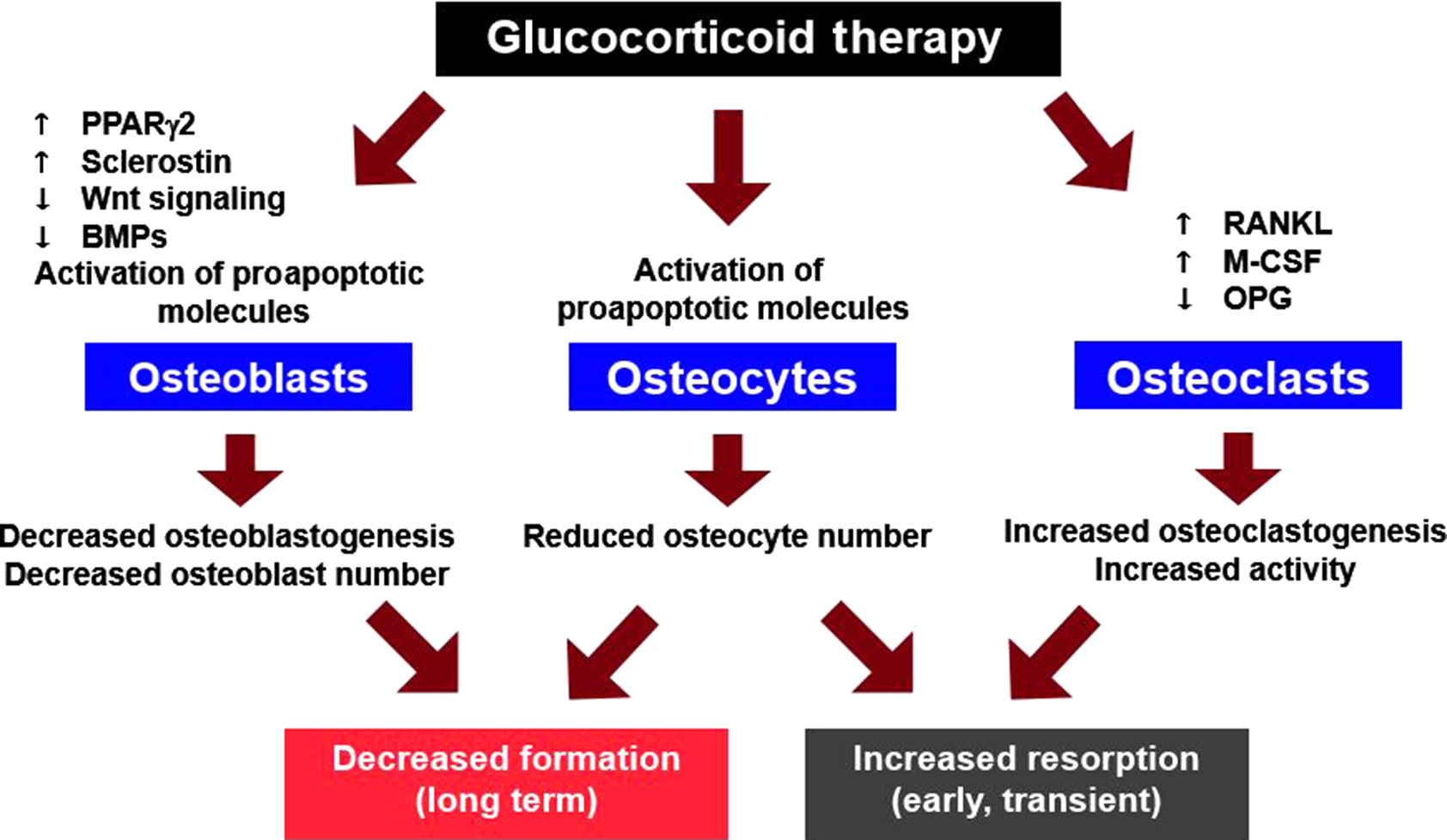
Although glucocorticoid excess has widespread systemic effects, the adverse skeletal impact is primarily due to direct action of these steroids on bone cells ( Fig. 45.4 ) . Glucocorticoids exert their effects by freely diffusing across cell membranes and binding to glucocorticoid receptors (GRs), which then release heat shock proteins. Subsequently, ligand-bound GRs may enter the nucleus, undergo dimerization, and regulate gene expression by attaching to glucocorticoid response elements in the promoter region of target genes, leading to altered protein synthesis and cell life span. GRs may remain monomeric in the cytoplasm and interact with transcription factors such as activator protein (AP)-1 and nuclear factor kappa B (NF-κB) to exert rapid and nongenomic effects on phagocytosis, neurophysiological function, and behavioral mechanisms . However, patients have been reported with osteonecrosis, osteoporosis, and Cushing’s syndrome due to treatment with low-dose glucocorticoids because of variations in GR number or sensitivity .
Prereceptor control of glucocorticoid action occurs through the 11β-hydroxysteroid dehydrogenase (11β-HSD) shuttle. Two isoenzymes of 11β-HSD (11β-HSD1 and 11β-HSD2) catalyze the interconversion of hormonally active glucocorticoids (e.g., cortisol) and inactive glucocorticoids (e.g., cortisone). The 11β-HSD1 enzyme is an activating route and the 11β-HSD2 enzyme is an inactivating route. Binding to the GRs and biological activity of any glucocorticoid depend on the presence of a hydroxyl at C-11. Therefore any tissue expressing 11β-HSDs can regulate the exposure of the cells in that tissue to active glucocorticoids . However, natural and synthetic glucocorticoids differ in their vulnerability to 11β-HSD2. With dexamethasone the 11-hydroxyl is already present as it is in prednisolone, but the fluorine atom at the 9α position of the B ring both extends the potency and occludes the 11β location. Therefore in contrast to prednisolone, dexamethasone could be invulnerable to inactivation by 11β-HSD2 and dexamethasone causes more skeletal complications than prednisone . Surprisingly, both aldosterone and cortisol have similar affinity for the mineralocorticoid receptor (MR) but serum total aldosterone levels are 100–400 pmol/L, whereas serum total cortisol circulates at 200–700 nmol/L. The 11β-HSD shuttle is necessary to protect the MR from the 1000-fold higher circulating concentration of cortisol. Physicians have long been perplexed by the occasional patient who develops clinical manifestations of Cushing’s syndrome with moon facies, buffalo hump, violaceous striae, central adiposity, hypertension, and diabetes when treated with relatively small doses of glucocorticoids, whereas other patients appear to be remarkably resistant to oral glucocorticoids. The sensitivity to exogenous glucocorticoids may be mediated by GR polymorphisms or inherited gradations in the 11β-HSD shuttle. This remarkable shuttle is a natural prereceptor controller of corticosteroid action as well as a unique tool that can be used to distinguish the direct effects of excess glucocorticoids on bone cells from the indirect effects that occur in almost every other tissue.
45.3.2
Direct effects of glucocorticoids on bone
Glucocorticoid-induced bone disease may be mediated by direct actions on bone cells, actions on extraskeletal tissues, or both. An animal model of GIOP is required to investigate this issue. The mouse is a faithful model of glucocorticoid-induced bone disease consistently demonstrating greater axial than appendicular bone loss accompanied by histological indices of impaired osteoblast function, decreased wall width, and increases in apoptotic osteoblasts and osteocytes, thus reproducing the major features of the human disease . To distinguish the direct from the indirect effects of glucocorticoids on bone, O’Brien et al. overexpressed 11β-HSD2 in transgenic mice utilizing the murine osteocalcin gene 2 (OG2) promoter, which is active only in mature osteoblasts and osteocytes. Using this promoter, the transgene did not affect normal bone development or turnover as demonstrated by identical BMD, strength, and histomorphometry in adult transgenic and wild-type animals, suggesting that endogenous glucocorticoid action in osteocalcin-expressing cells is not required for normal skeletal development. Additional strong evidence against a beneficial role for endogenous glucocorticoids on bone in adults is supplied by reports of normal BMD values in patients with Addison’s disease until they are treated with greater than replacement amounts of glucocorticoids . Patients receiving glucocorticoids for adrenal insufficiency are taught to increase doses when stressed by an intercurrent illness. Misinterpretation of such teaching may also lead to excess dosing and decreases in BMD .
Wild-type mice receiving excess glucocorticoids showed the expected decreases in osteoblastogenesis in the bone marrow and numbers of osteoblasts on cancellous bone, diminished osteoid and bone formation, and increased apoptosis of osteoblasts and osteocytes typical of the clinical bone disease . However, mice harboring the OG2–11β-HSD2 transgene were protected from glucocorticoid-induced apoptosis of osteoblasts and osteocytes. Prevention of osteoblast/osteocyte apoptosis, in turn, resulted in the preservation of cancellous osteoblast numbers and osteoid production, thereby preventing the expected decrease in bone formation caused by administration of excess glucocorticoids. More strikingly, bone strength was preserved in the transgenic mice despite loss of BMD, suggesting that osteocyte viability independently contributed to bone strength.
Using the same approach, these workers overexpressed 11β-HSD2 in transgenic mice utilizing the tartrate-resistant acid phosphatase (TRAP) promoter that is active only in osteoclasts. Again, morphometric measurements of body size, vertebral and femoral dimensions, and vertebral histomorphometry indicated that the transgene had no impact on normal skeletal development. When the animals were challenged with prednisolone, there were similar increases in the prevalence of osteoblast apoptosis and decreases in osteoid area, osteoid perimeter, osteoblast number, and bone formation rate in both wild-type and transgenic mice. The seminal observation was that glucocorticoid administration dramatically reduced the osteoclast number in the transgenic mice but not in the wild-type animals, in which glucocorticoids promote osteoclast survival . Furthermore, there was a greater than fourfold loss of spinal BMD in the wild-type mice compared to the placebo animals, but in the transgenic animals, the prednisolone-induced bone loss was abrogated . These results provide strong in vivo evidence that osteoclasts are direct targets of glucocorticoid action and that this direct action prolongs the life span of these cells. Moreover, the evidence indicates that the direct action of glucocorticoids on osteoclasts is the major cause of the early, rapid loss of bone in states of glucocorticoid excess.
45.3.3
Effects of glucocorticoids on osteoblast differentiation and osteoclast proliferation
Glucocorticoids reduce osteoblast differentiation by attenuating Akt (protein kinase B) phosphorylation and increasing activation of the redox-sensitive forkhead box subgroup O transcription factor family (FoxOs), which in turn inhibit wingless (Wnt)/β-catenin signaling—a critical pathway for the generation of osteoblasts . Glucocorticoids also enhance the expression of Dickkopf-1, an antagonist of the Wnt pathway and suppress bone morphogenetic proteins, factors required to induce osteoblast differentiation . In addition, glucocorticoids also increase production of peroxisome proliferator-activated receptor γ, a transcription factor that induces terminal adipocyte differentiation while suppressing osteoblast differentiation, potentially contributing to increased marrow fat and reduced osteoblasts. Animals fully deficient in sclerostin, an inhibitor of Wnt-mediated bone formation, have considerable resistance to the effects of glucocorticoids on bone .
Osteoclast differentiation and survival are regulated by the receptor activator of NF-κB ligand (RANKL), a member of the tumor necrosis factor ligand family. RANKL and macrophage colony-stimulating factor are sufficient for osteoclast differentiation, and RANKL also prolongs the survival of differentiated osteoclasts.
Glucocorticoids downregulate the mRNA for osteoprotegerin (OPG), a soluble decoy receptor for RANKL, while these drugs increase RANKL mRNA in preosteoblastic cells . Decreased levels of OPG could permit RANKL to increase osteoclastogenesis by unopposed binding to its specific receptor, RANK, on the surface of hematopoietic osteoclast progenitor cells. The resultant decrease in the OPG/RANKL ratio could then lead to an increase in the number of cancellous osteoclasts on bone and explain the early, rapid bone loss typical of GIOP. However, other studies have shown that exogenous glucocorticoids do not alter RANKL or OPG mRNA levels in vivo . Moreover, recent work indicates that RANKL produced by osteoblasts or their progenitors does not contribute to bone remodeling in mature animals and that osteocytes are the major RANKL-producing cells that control osteoclast formation .
45.3.4
Effects of glucocorticoids and alterations in sex hormones
Glucocorticoids are known to affect the hypothalamic–pituitary–gonad axis, probably at several points. In a study of normal men by Pearce et al. , prednisolone decreased serum levels of total testosterone, estradiol, and adrenal androgens such as androstenedione and dehydroepiandrosterone sulfate. Sex hormone–binding globulin (SHBG) levels also decreased, making the ratio of testosterone to SHBG unchanged after glucocorticoid administration. On the other hand, this ratio is considered to be less robust than measurements of bioavailable testosterone, calculated by mass action equations from the total testosterone, SHBG, and albumin levels. In both women and men , bioavailable estradiol is correlated with BMD. In a study by Fink et al. , bioavailable estradiol was clearly associated with BMD in a large group of older men in a prospective trial (MrOS). In earlier studies , glucocorticoids were shown to decrease secretion of sex hormones at various levels. If glucocorticoids can alter sex steroids, then such effects may, in turn, change calcium metabolism. For example, decreased estrogen may lead to increased urinary calcium excretion. In an earlier study , BMD and plasma estradiol were correlated in women taking oral glucocorticoids. In a study by Lane et al. , hormone replacement alone was able to maintain BMD in postmenopausal women on oral glucocorticoids.
In men, testosterone administration provides both androgen and estrogen because some of the testosterone is converted to estradiol. Thus in the study by Reid et al. , testosterone was able to improve BMD in men with COPD treated with oral glucocorticoids. In another study , testosterone and nandrolone that are not converted to estradiol were administered to men receiving an average dose of approximately 13 mg of prednisone daily. Only in the men receiving testosterone was there an increase in spine BMD, whereas both androgens increased muscle mass. This study suggests that androgens must be aromatized to estrogens in order to have a salutary impact on bone in glucocorticoid-treated men. Bone turnover markers were no different among men treated with testosterone, nandrolone, or placebo.
In summary, although sex hormone replacement has a salutary effect on patients with GIOP, there is little evidence of a significant impact on calcium metabolism. Testosterone can increase muscle strength and size. Thus improvements in BMD in men treated with androgens may be related to indirect effects.
Comparison of the effects of hypogonadism and glucocorticoid excess on bone cells provides further insight into the potential aggravation of glucocorticoid-induced bone loss by decreased sex steroids. The loss of gonadal function in either sex stimulates the production of osteoblasts and osteoclasts in the bone marrow, resulting in an increase in cancellous osteoblasts, osteoclasts, and bone turnover—changes that are quite distinct from those found in GIOP ( Table 45.1 ) . Nonetheless, the histological findings in GIOP have been attributed in part to secondary hypogonadism . As described previously, Pearce et al. found a 4.6% decrease in spinal BMD after 6 months of 50 mg daily of prednisone to suppress antisperm antibody formation in infertile men, despite the maintenance of a normal testosterone/SHBG ratio as well as restoration of fertility. Additional strong evidence against the concept that hypogonadism is universal in glucocorticoid-treated patients is provided by registries containing the records of thousands of successful pregnancies in woman receiving prednisone to prevent rejection of renal transplants . Furthermore, in several studies , seminal vesicle weight was not decreased when prednisolone was administered to mice. Moreover, in an animal model of GIOP, hypogonadism does not occur in, or contribute to GIOP and the adverse effects of glucocorticoids override those of hypogonadism . In addition, amenorrheic and eumenorrheic women with Cushing’s syndrome have similar BMD . The adverse effects of sex steroid withdrawal on bone are mediated by stromal osteoblastic cells and are obviated by constraints on osteoblastogenesis whether genetically defective, as in senescence-accelerated mice (SAMP6), an animal model of defective osteoblastogenesis , or acquired because of glucocorticoid excess. Some investigators have reported that addition of glucocorticoids to cultured osteoprogenitor cells increases osteoblastic cell differentiation, but this in vitro phenomenon is inconsistent with the profound decrease in osteoblasts and bone formation that occurs when pptatients are exposed to glucocorticoid excess .
| In cancellous bone |
| Marked reduction in bone area with decreased trabecular width |
| Diminished wall width |
| Decreased osteoid area |
| Decreased numbers of osteoblasts |
| Increased prevalence of osteoblast and osteocyte apoptosis |
| Normal or slightly increased numbers of osteoclasts |
| Prolongation of the reversal phase |
| Decreased rate of bone formation |
| In cortical bone |
| Increased cortical porosity |
| Increased prevalence of osteocyte apoptosis |
| Decreased rate of bone formation |
45.3.5
Effect of glucocorticoids on absorption of calcium from the gut and on metabolism of vitamin D
Although studies show that glucocorticoids decrease gut absorption of calcium, there is question as to whether altered vitamin D levels or effects might be involved . Part of this confusion may relate to the fact that vitamin D insufficiency, in general, is common . Thus the patient started on glucocorticoid therapy may already have lower circulating 25-hydroxy (OH) vitamin D levels, and any direct effect of glucocorticoids on the gut will be influenced by extant vitamin D status. Again referring to the study of Pearce et al. , in which otherwise normal men were given prednisolone, there was no decrement in serum calcium or increase in serum parathyroid hormone (PTH), suggesting that although there may be decreased gut absorption of calcium, it does not play a major role in the pathogenesis of GIOP.
45.3.6
Glucocorticoid effects on renal calcium and phosphate handling
In a study of patients with multiple sclerosis receiving large doses of glucocorticoids , the serum phosphate level decreased as urinary excretion of phosphate increased on the first day after the start of therapy. Interestingly, in this study, there was little change in urinary calcium excretion acutely, but with more chronic therapy, the urinary calcium increased. With time, both urinary calcium and phosphate excretion returned toward normal. There may be direct effects on renal tubular calcium handling or an increased filtered load of calcium due to mobilization of skeletal calcium. In addition, the underlying disorder for which glucocorticoids are prescribed may have an effect on mineral excretion . In sarcoidosis, for example, there may be a state of increased calcitriol.
45.3.7
Effect of glucocorticoids on parathyroid hormone secretion and activity
For many years, it has been postulated that stimulation of PTH secretion by metabolic alterations caused by glucocorticoid excess might play a role in GIOP. For example, decreased gut absorption of calcium and increased urinary calcium excretion have been suggested as stimuli to secondary hyperparathyroidism, presumably via an imperceptible fall in the serum calcium level . In recent studies, evidence for increased PTH secretion has been less convincing. In the study by Pearce et al. , normal men received 50 mg of prednisolone daily to reduce antisperm antibodies. The men were completely normal in every other way, in contrast to many previous studies. Despite the large doses of prednisolone, there was no decrease in the serum calcium nor was the serum PTH elevated. Thus as reviewed by Rubin and Bilezikian , it is unlikely that secondary hyperparathyroidism due to glucocorticoid effects plays an important role in the pathophysiology of GIOP.
In a study comparing men on chronic prednisone therapy with a control group of men, serum PTH levels were measured every 3 minutes for 6 hours . The men on glucocorticoids had a low PTH tonic rate but higher fractional pulsatile secretion, leading to similar overall PTH concentration and mean integrated area under the curve in the control and prednisone-treated men.
It has also been postulated that glucocorticoid-induced bone resorption is mediated by secondary hyperparathyroidism. Evidence for this has been called into question . Elevated concentrations of PTH are not typical of patients receiving glucocorticoid therapy; levels of bone resorption markers, such as the urinary excretion of the N -telopeptide of type I collagen, show either no change or a decrease with long-term glucocorticoid therapy; and bone histomorphometry in GIOP shows marked suppression of osteoblasts and bone formation ( Table 45.1 ), in contrast to the augmented bone turnover typical of hyperparathyroidism .
45.3.8
Histomorphometry of glucocorticoid-induced osteoporosis
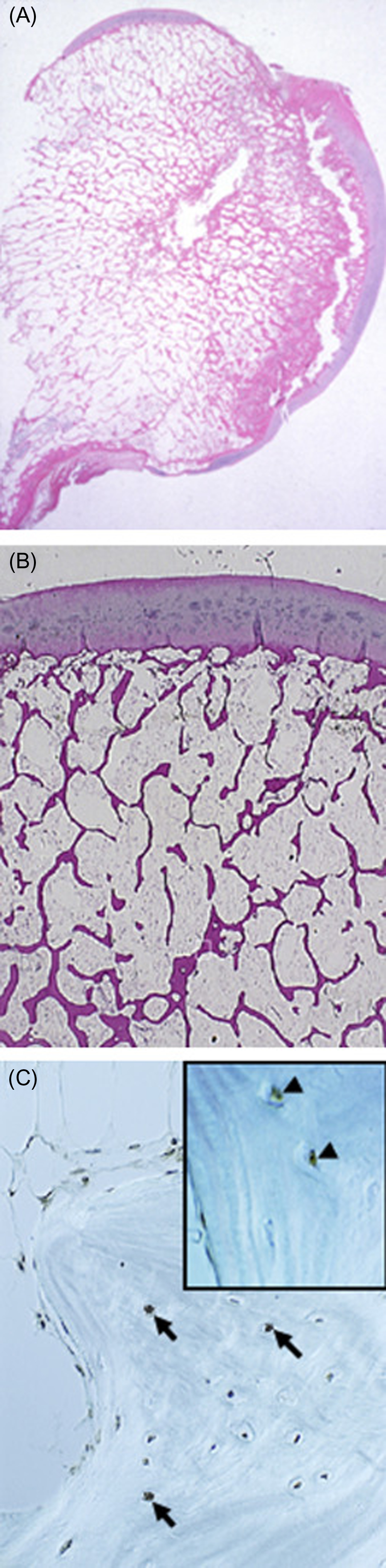
The number of osteoblasts in the basic multicellular unit of bone is a crucial determinant of the rate of bone formation. Osteoblast number, in turn, depends on the balance between the supply of new cells, reflecting the replication and differentiation of osteoblast progenitors, and on the life span of mature osteoblasts, reflecting the timing of their death by apoptosis. Histomorphometric studies in patients receiving long-term glucocorticoid treatment consistently show reduced numbers of osteoblasts on cancellous bone and diminished wall width, a measure of the work performed by these cells . The decreased osteoblasts are due to glucocorticoid-induced reductions in the production of new osteoblast precursors as well as to premature apoptosis of mature, matrix-secreting osteoblasts ( Table 45.1 and Figs. 45.5 and 45.6 ) . The increased apoptosis typical of GIOP thus markedly depletes the already suboptimal pool of osteoblasts. Therefore it should not be surprising that decreases in trabecular width, osteoid area, wall width, and bone formation rate are common histological findings in glucocorticoid-treated patients . Normalization of these findings and prevention of osteoblast and osteocyte apoptosis in glucocorticoid-treated transgenic mice that overexpressed 11β-HSD2 exclusively in mature osteoblasts and osteocytes clearly show that the adverse effects of excess glucocorticoids directly on these mature cells predominate over their effect on progenitors in vivo . Inadequate numbers of osteoblasts are also an important cause of the reduction in cancellous bone area and decrease in trabecular width, a result of incomplete cavity repair during bone remodeling . With glucocorticoid excess, cancellous bone area is often less than 12% and correlates with the decreased wall width . Cortical bone demonstrates increased porosity, whereas cortical width ranges from clearly subnormal to within normal limits, at least at the iliac crest .

Some clinical histomorphometric studies of GIOP have reported moderate increases in the erosion perimeter, but others have shown no significant change. Increased erosion perimeter may, however, represent only accumulation of the reversal perimeter, erosion cavities devoid of osteoclasts and accumulating merely because of delayed bone formation . When carefully measured, osteoclast numbers in patients receiving chronic glucocorticoid treatment are within the normal range or just slightly above normal . The differences found in the clinical studies are due, in part, to the time at which the bone biopsy was obtained. Some biopsies were collected within the first 5–7 months of treatment, when an early and transient increase in osteoclasts has been demonstrated, whereas others were obtained after more than 5 years of glucocorticoid therapy, when the numbers of both osteoclasts and osteoblasts are profoundly reduced. Prevention of the increase in osteoclast numbers and bone resorption in glucocorticoid-treated transgenic mice overexpressing 11β-HSD2 exclusively in mature osteoclasts clearly shows that the adverse effects of excess glucocorticoids on bone resorbing cells are also primarily direct .
Aging and glucocorticoid excess cause a decline in bone strength that is greater than the decline in bone mass in humans and mice and both conditions reduce bone fluid, skeletal blood flow, and vascular endothelial growth factor or VEGF. Along these lines, aging in humans and mice is associated with an increase in adrenal production of glucocorticoids as well as bone expression of 11β-HSD1, a marker of increased endogenous glucocorticoids . Aging in mice also decreased the volume of the bone vasculature and solute transport from the peripheral circulation to the lacunar–canalicular system. Furthermore, mice with osteoblast- and osteocyte-specific transgenic expression of 11β-HSD2 were protected from the adverse effects of aging on osteoblast and osteocyte apoptosis, bone formation rate and microarchitecture, crystallinity, vasculature volume, interstitial fluid, and strength. Thus endogenous glucocorticoids also contribute to the loss of bone strength with aging.
45.4
Other musculoskeletal effects of glucocorticoids
45.4.1
Osteonecrosis
Glucocorticoid administration is often overlooked as the most common cause of nontraumatic osteonecrosis. Glucocorticoid-induced osteonecrosis develops at the hip, shoulder, knee, or ankle in 9%–40% of patients receiving long-term therapy, although it may also occur with short-term exposure to high doses, after intraarticular injection, and without GIOP . The mechanism underlying glucocorticoid-induced osteonecrosis has been postulated to be increased marrow fat resulting in an increase in intraosseous pressure and a decrease in bone perfusion, fat embolism, and hypercoagulability reducing blood flow to the femoral head, accumulation of fatigue fractures, and osteocyte apoptosis .
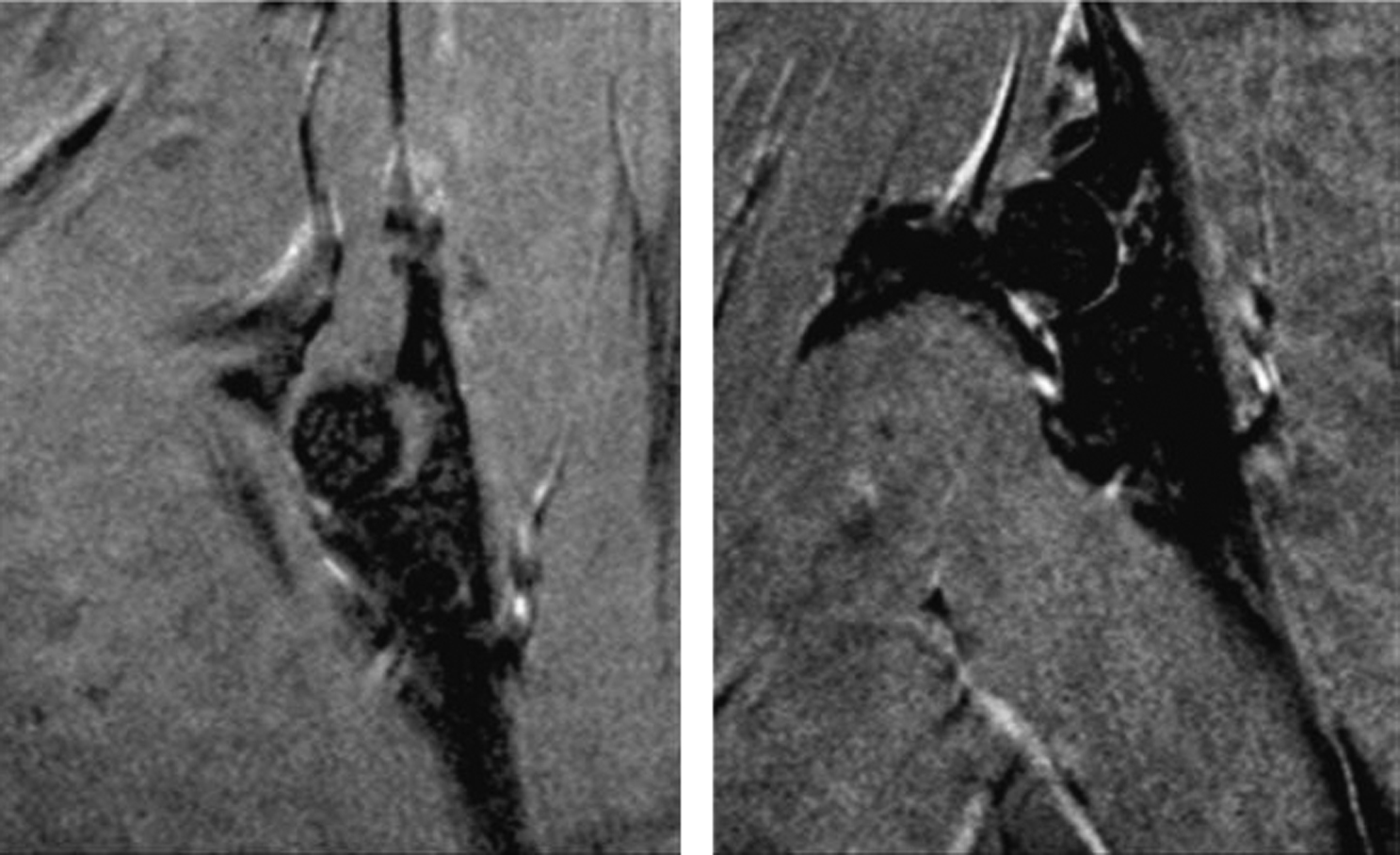
Recent findings have highlighted the pathophysiology of glucocorticoid-induced osteonecrosis . Shortly after implantation of slow-release prednisolone pellets in a murine model of osteonecrosis, the femoral head, but not the distal femur, showed a decrease in the expression of the hypoxia-inducible factor (Hif)-1a and VEGF, the number of osteoblasts, and bone formation rate and strength and showed an increase in osteoclasts. These changes were accompanied by conversion of the normal dendritic vasculature to pools of edema as detected by magnetic resonance imaging, providing diagnostic evidence of early osteonecrosis ( Fig. 45.7 ). During the time period with the early histologic findings, there were no detectable changes in bone density, cortical or cancellous bone architecture, mid-shaft or distal cancellous bone, or osteocyte apoptosis. Later in the disease course, femoral head cancellous density, cortical width, and trabecular thickness decreased, and the femoral heads had full-depth cortical penetrations and cancellous tissue osteonecrosis. Because of the weight that it bears and inadequate vascular supply, the femoral head is a particularly sensitive anatomical site to the adverse effects of glucocorticoid excess on bone. Decreases of Hif-1a and VEGF expression, bone vascularity, and strength precede the loss of bone mass and micro-architectural deterioration, thus rendering the femoral head vulnerable to collapse ( Fig. 45.8 ).
45.4.2
Glucocorticoid-induced myopathy
Loss of muscle mass is considered a cardinal sign of clinical glucocorticoid excess, Cushing’s syndrome. The classic patient has central obesity and thin limbs. At least part of this is due to the metabolic effect of glucocorticoids, increasing gluconeogenesis by providing substrate in the form of amino acids from protein breakdown in muscle .
Studies suggest multiple other mechanisms for the loss of muscle in states of glucocorticoid excess. For example, there may be loss of thick myosin filaments from muscle, suggesting decreased transcription rate of myosin . In studies of rat muscle treated with dexamethasone, other enzymes also appeared to affect myosin loss. The sodium–potassium pump of muscle may also be affected by glucocorticoids , leading to weakness in some muscles. In dogs, spontaneous pituitary-dependent Cushing’s disease is accompanied by decreased sodium–potassium ATPase content, leading to decreased exercise endurance .
In an animal model of glucocorticoid myopathy, a report suggested that hydroxyl radical may mediate muscle weakness due to glucocorticoids. Other studies have focused on the mechanism of skeletal muscle apoptosis in animal models of glucocorticoid myopathy. Muscle atrophy due to glucocorticoids may be mediated by augmented expression of E3 ubiquitin ligases (Atrogin-1 and MuRF-1). A recent study suggests that the 11β-HSD1 enzyme is responsible for regulation of these ubiquitin ligases.
The effect of muscle contractions on bone strength is discussed elsewhere (see Chapter 15 : Adaptation of skeletal structure to mechanical loading, and Chapter 23 : Reproductive and hormonal factors and the risk for osteoporosis), but suffice it to say that lack of skeletal loading leads to bone loss. Examples of this are spinal cord injury, immobilization, and low-gravity states. Thus in glucocorticoid excess, the loss of muscle by whatever mechanism translates to less skeletal loading and potentially contributes to the osteoporosis of endogenous or exogenous Cushing’s syndrome. In a study by Natsui et al. , high-dose glucocorticoid treatment for 2 months resulted in demonstrable loss of both bone mineral content and lean body mass.
In patients treated with glucocorticoids for inflammatory diseases, clinical improvement may result in increased exercise tolerance, off-setting the deleterious effects on muscle. In a study of heart transplant patients treated with glucocorticoids to prevent rejection , resistance exercise prevented changes in skeletal muscle phenotype observed with glucocorticoid use. In a recent study in prednisolone-treated rats , alfacalcidol preserved muscle volume and strength.
45.5
Effects of Cushing’s syndrome on bone
In his landmark 1932 paper, Cushing described an autopsy performed on a patient with a basophilic pituitary tumor . He found, “… skeletal osteoporosis with spinal curvature, the bones being easily cut with a knife”. All forms of Cushing’s syndrome affect the skeleton . More than 75% of patients with the syndrome have fractures, regardless of the etiology of the hypercortisolism. Although it was once thought that adrenocorticotrophic hormone (ACTH) stimulation of the adrenal production of dehydroepiandrosterone (DHEA), androstenedione, and testosterone could have protective effects on the skeleton, evidence suggests otherwise. In 80 consecutive patients with Cushing’s syndrome of varying etiologies, the lowest BMD values were found in patients with ectopic ACTH secretion and in these patients, multiple vertebral fractures were most frequent. In addition, BMD values and fracture prevalence was equivalent in amenorrheic and eumenorrheic women with Cushing’s syndrome.
GIOP is much more often caused by exogenous systemic glucocorticoids. However, endogenous glucocorticoid excess (Cushing’s syndrome) also leads to osteoporosis in a substantial number of patients. Endogenous Cushing’s syndrome can be due to overproduction of cortisol by a primary adrenal process (an adenoma or carcinoma) or more likely by excess production of ACTH by a pituitary adenoma or by a neoplasm (usually a pulmonary small cell carcinoma or a neuroendocrine tumor). Unlike Cushing’s syndrome due to systemic glucocorticoid administration, the impact on bone from the concurrent glucocorticoid required to treat an inflammatory disorder, such as RA or chronic bronchitis, is not likely present in patients with endogenous glucocorticoid excess. It is more likely that only the effects of augmented glucocorticoid secretion on bone may be found in endogenous Cushing’s syndrome. However, some adrenal tumors in women may secrete androgens as well as glucocorticoids, causing hirsutism and other signs of excess androgen. Whether this second hypersecretion helps to mitigate the deleterious impact of the glucocorticoid excess is difficult to determine. Excess mineralocorticoid may also affect bone. Another subgroup of adrenal causes of Cushing’s syndrome is bilateral nodular hyperplasia, which may result in variable amounts of hormone excess from the three layers of the adrenal cortex. The unregulated hypersecretion of ACTH, particularly by small cell lung cancer, may markedly stimulate all layers of the adrenal gland resulting in cortisol, androgens, and aldosterone to be secreted in great excess. The patient with ectopic ACTH syndrome may also suffer from the systemic manifestations of lung or other cancer, including cachexia, that may also affect bone. Pituitary tumors cause ACTH excess to a much smaller degree than small cell lung cancer, but the augmented glucocorticoid secretion may be complicated by hypogonadism (due to decreased secretion of gonadotropins) and decreased growth hormone secretion. Thus endogenous glucocorticoid excess may be accompanied by other hormonal and systemic abnormalities, which also affect bone and fracture risk, including glucocorticoid-induced myopathy. Nonetheless, it is important to note that patients with endogenous glucocorticoid excess are at high risk for osteoporosis and fracture. In one study the prevalence of vertebral fracture in a group of patients with various forms of endogenous cortisol excess was 76%. Those patients with ectopic ACTH had the highest cortisol levels and the greatest frequency of multiple vertebral fractures. In other reviews of the subject, the overall estimate of fractures was high, with 30%–50% of patients having vertebral fractures . Other bones may be affected as well. One study found that even lower extremity fractures may be observed in endogenous Cushing’s syndrome . While trabecular bone is clearly affected by endogenous glucocorticoid excess, as manifested by the high prevalence of vertebral fractures, cortical bone may also be involved. High-resolution peripheral quantitative computed tomography (HR-pQCT) can be used to assess the distal radius and distal tibia . In both the radius and tibia, the cortical area, cortical thickness, and cortical density were lower in Cushing’s patients. In addition, volumetric BMD was lower in the tibia in the patients, compared to control subjects.
The most interesting and controversial cause of Cushing’s syndrome is that known as subclinical Cushing’s syndrome or mild autonomous cortisol secretion. Subclinical Cushing’s syndrome may be diagnosed in some patients who have incidentally discovered adrenal neoplasms, incidentalomas. The patients with adrenal adenomas that appear to make glucocorticoids to a slight excess are at higher risk for fracture. The definition of subclinical Cushing’s syndrome is determined by cortisol values after dexamethasone suppression that are in a gray zone between normal and definitely abnormal. It is difficult to determine the risk of fracture of subclinical Cushing’s syndrome compared to patients with overt endogenous Cushing’s syndrome, but in patients with adrenal incidentaloma, those with subclinical Cushing’s syndrome had more vertebral fractures than those with completely normal cortisol secretion .
Some patients with endogenous glucocorticoid excess may first come to medical attention because of a vertebral fracture. The actual incidence of fracture is very high in patients with endogenous Cushing’s syndrome, but it varies widely in reported series. BMD testing by dual-energy x-ray absorptiometry (DXA) is also variable, but most patients will meet criteria for osteopenia or osteoporosis. It should be assumed that every patient is at high risk for fracture, but almost all should be assessed by DXA and other means to quantitate the risk. Newer techniques have been used to improve fracture risk assessment in endogenous Cushing’s syndrome. As discussed earlier, HR-pQCT has provided evidence of abnormalities of cortical bone in addition to the well-known abnormalities of trabecular bone. Recently, trabecular bone score (TBS), a textural analysis of spine DXA measurements, has been studied in endogenous Cushing’s syndrome. TBS was used to evaluate patients with manifest endogenous Cushing’s syndrome, patients with subclinical Cushing’s syndrome, and patients with adrenal incidentaloma without mildly excess glucocorticoid levels . Low TBS scores were more common than osteoporosis by DXA in patients with endogenous Cushing’s syndrome. Among the patients with adrenal incidentaloma, those with mild cortisol excess had lower TBS scores but not lower BMD than incidentaloma patients with normal cortisol secretion. More studies are needed to further characterize the bones of patients with endogenous glucocorticoid excess, including those with the milder cases.
45.6
Treatment options and fracture risk reduction
Although almost 70 years have elapsed since glucocorticoids were first used to treat inflammatory diseases, the unparalleled short-term benefits of glucocorticoids in reducing inflammation and controlling symptoms have perpetuated their continued widespread use. Indeed, evidence supports a sustained, disease-modifying role for low-dose glucocorticoid administration in RA . However, high rates of associated adverse events, particularly fragility fractures, are commonly observed in patients receiving chronic glucocorticoid therapy , even when used at low doses . Therefore health-care providers need to carefully balance the desired benefits of glucocorticoid therapy against the potential toxicities, especially GIOP.
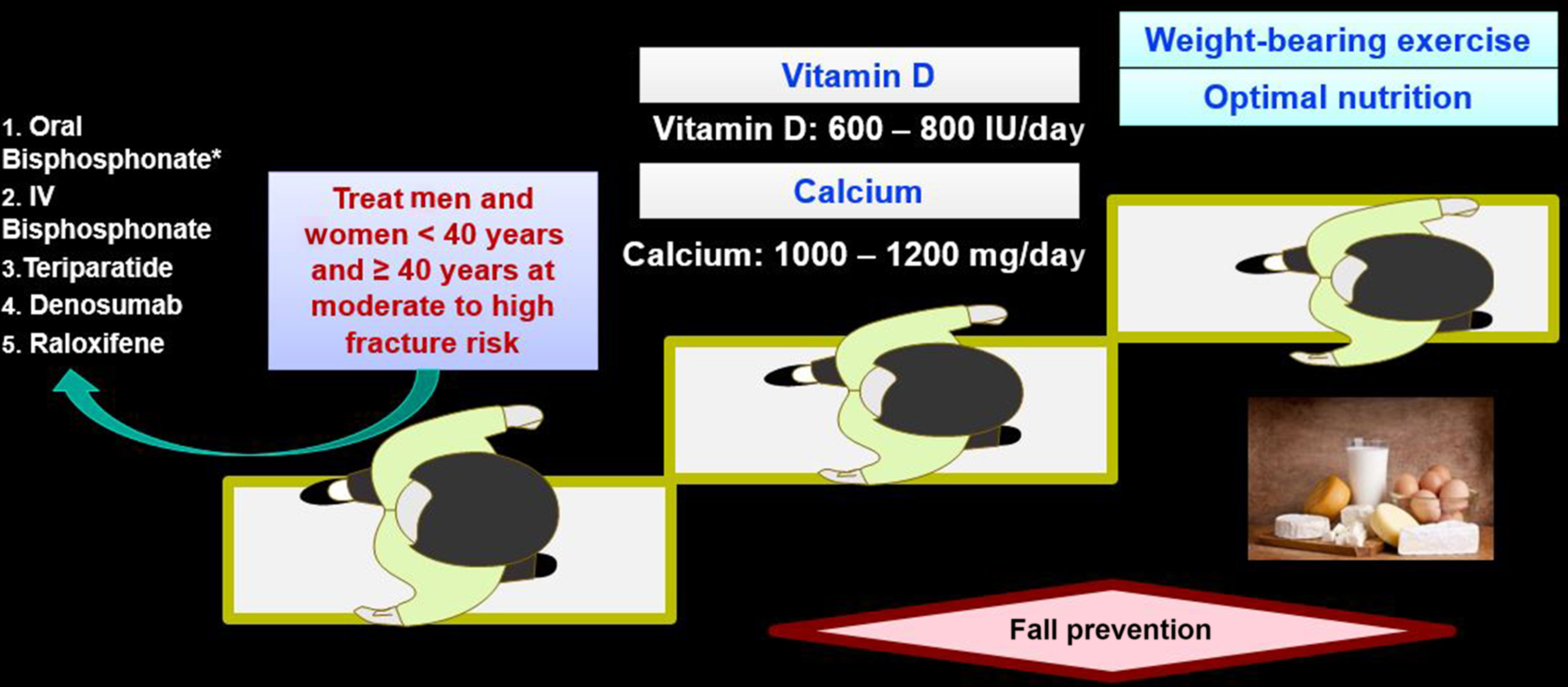
Lifestyle modification and both pharmacologic and nonpharmacologic measures to reduce the risk of GIOP are appropriate for all patients receiving long-term glucocorticoids. Although the definition of “long-term use” remains controversial, ongoing or expected use of greater than 3 months constitutes a reasonable threshold for concern . Dose reduction or complete cessation of glucocorticoid therapy is always desirable , but this may not be achievable for many patients. Nonpharmacologic interventions such as smoking cessation, weight-bearing exercise, reduction in alcohol intake, and interventions to mitigate fall risk should be provided to all patients at clinical risk for fracture. These recommendations are based on Level C evidence . Therapeutic doses of calcium and vitamin D are necessary but may not be sufficient for patients receiving chronic glucocorticoids. Of the several prescription medications that are used to prevent or treat GIOP, the oral bisphosphonates alendronate (approved for treatment of GIOP) and risedronate (approved for both prevention and treatment of GIOP), the intravenous bisphosphonate zoledronic acid (approved for treatment), and denosumab are Food and Drug Administration (FDA)-approved for use for GIOP in the United States. In addition, the anabolic agent teriparatide is also FDA-approved for GIOP. Regulatory agencies in other countries have also granted approval for these various agents. Furthermore, for management of GIOP, some biologic rationale and/or more limited scientific evidence exists for other nonapproved agents, including other parenteral bisphosphonates, calcitonin, sex hormones, selective estrogen receptor modulators (SERMs), activated vitamin D preparations, thiazide diuretics, and vitamin K. There is also strong biologic rationale for the use of two newer anabolic agents: abaloparatide (a PTH-related peptide analog) and romosozumab (a monoclonal antibody to sclerostin). Neither agent has been tested in large numbers in GIOP nor is there regulatory approval for this indication.
45.6.1
Calcium and vitamin D
All GIOP management guidelines advocate calcium and vitamin D supplementation . Recommended calcium doses are at least 1200 mg/day. Since the calcium content of foodstuffs in the average adult diet is insufficient to meet this target, for most glucocorticoid users, the extra dietary calcium needs to be consumed (preferred) or calcium needs to be given as oral supplements. As discussed in the Institute of Medicine (IOM) report on calcium and vitamin D , the American diet has increased calcium content, most likely due to the presence of more calcium-fortified foods. The IOM recommends that adults up to age 70 consume a total of 1000 mg of elemental calcium daily and people over age 70 consume 1200 mg daily. However, calcium alone has only a modest beneficial effect on bone turnover in glucocorticoid-treated patients , and calcium monotherapy is insufficient to prevent or treat GIOP . In conjunction with calcium, a variety of vitamin D preparations, including ergocalciferol, cholecalciferol, and activated forms of vitamin D such as calcitriol, are available. Studies supporting recommendations for calcium and vitamin D supplementation in GIOP include a 2-year trial of 65 RA patients treated chronically with low-dose prednisone (approximately 5 mg/day) randomized to 1000 mg of calcium carbonate and 500 IU of ergocalciferol versus placebo . Those given the daily supplements gained 0.7% and 0.9% annually in lumbar spine and trochanter BMD, respectively, compared to losses of −2.0% and −0.9% at these sites in the placebo group. Data from the placebo arm of bisphosphonate clinical trials also demonstrated relative BMD preservation, more so in the spine than the hip, in patients receiving daily calcium and inactive vitamin D . Many of the patients in these clinical trials were at lower risk of fractures because of premenopausal status or having normal BMD at study entry.
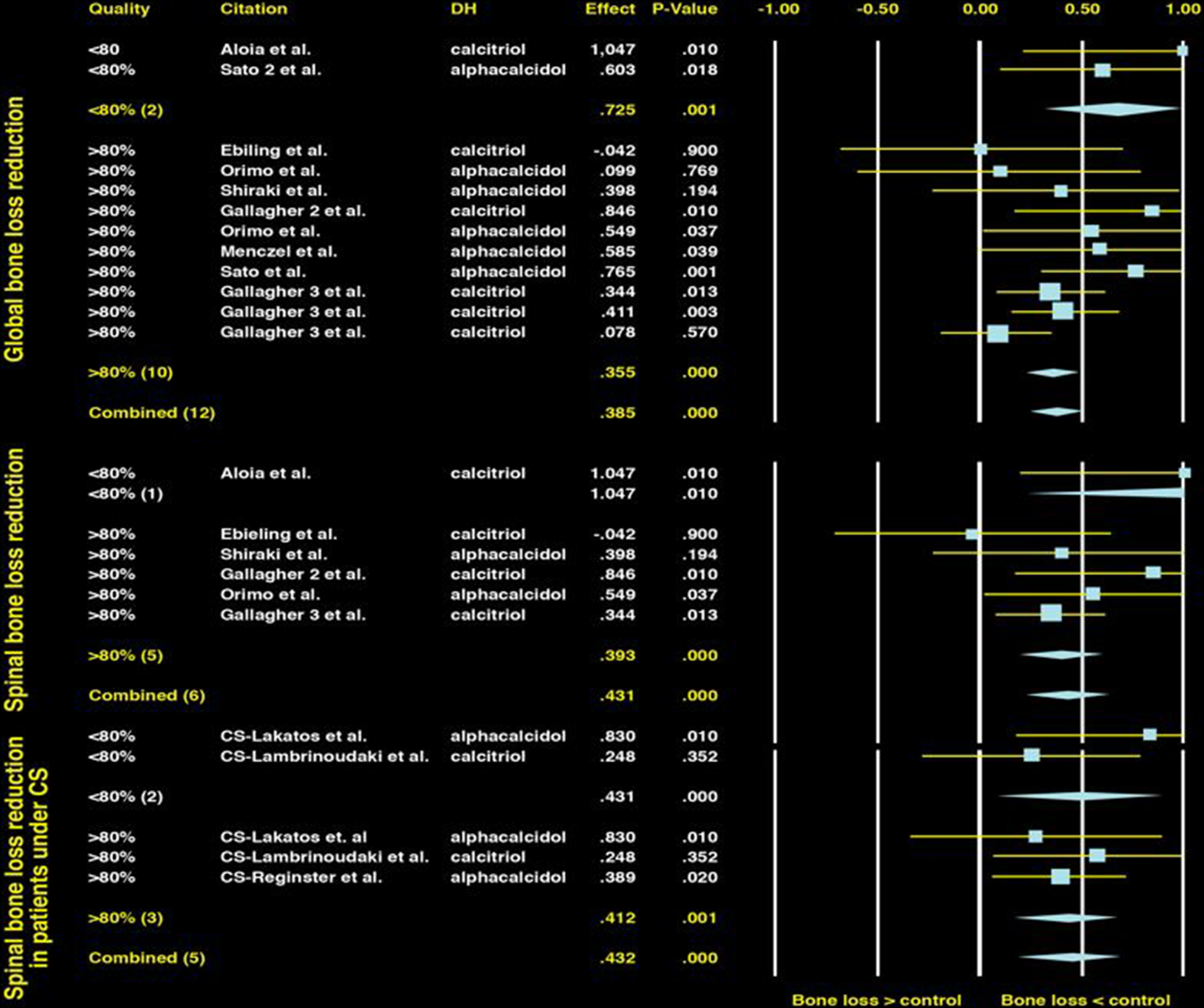
Most but not all studies of native vitamin D (cholecalciferol or ergocalciferol) or active vitamin D metabolites (e.g., calcitriol and alfacalcidol) have shown preservation or only modest BMD losses in glucocorticoid-treated patients . The results of many of these studies have been summarized in metaanalyses of vitamin D, which have demonstrated consistent findings ( Fig. 45.9 ). In one metaanalysis , both active vitamin D analogues and native vitamin D were able to maintain lumbar spine BMD similarly to one another and significantly better than no treatment (effect size, 0.38; P <.001 and .41 and P =.002, respectively). There are few direct comparisons between active and native vitamin D . However, one study demonstrated significant preservation of BMD at the lumbar spine (2.4% vs −0.8%, P <.0001) and a significant reduction in vertebral fractures over 3 years in patients treated with alfacalcidol compared to cholecalciferol (vertebral fracture rate difference, 15%; 95% CI, 7–25) . Hypercalcemia was uncommon in both the active and the native vitamin D groups and occurred in <3% of patients.
Metaanalyses that pooled small trials showed that vitamin D was inferior to the newer generation amino-bisphosphonates . To support and extend this observation, one study randomized 201 rheumatic disease patients receiving 7.5 mg of prednisone per day to either alendronate (10 mg daily) or alfacalcidol (1 µg daily) . These individuals (mean age, 61 years) had relatively normal BMD at the beginning of the trial (lumbar spine and total hip T scores >−1.0). At 18 months the lumbar spine BMD in the alendronate group had increased by 2.1% (95% CI, 1.1%–3.1%) and it had decreased by 1.9% (95% CI, −3.1% to −0.7%) in the alfacalcidol group. Three patients in the alendronate group had one new vertebral deformity compared to eight patients with a total of 13 new vertebral deformities in the alfacalcidol group (hazard ratio, 0.4; 95% CI, 0.1–1.4). The absolute fracture risks were approximately 3% versus 8% over 18 months. To prevent one vertebral fracture over this time period, 20 patients would have to be treated with alendronate. Similar results, preferentially favoring alendronate over alphacalcidol on BMD, were seen in a study targeting patients with inflammatory bowel disease. Alendronate was also well tolerated in those patients .
Regardless of the preparation of vitamin D used, high fracture rates were observed in most of these studies. This was especially true in patients older than age 50 years or with previous fragility fractures. For the majority of individuals, calcium and vitamin D supplementation is necessary but not sufficient to reduce GIOP-related fracture morbidity. Even active vitamin D metabolites will have limited efficacy to reduce bone loss in patients treated with moderate- or high-dose glucocorticoids. Experts suggest measuring serum levels of 25-(OH) vitamin D and repleting patients with osteoporosis with amounts adequate to raise the serum level to at least 30 ng/mL (see Chapter 71 : Nutrients beyond calcium and vitamin D to treat osteoporosis). For the population at large the IOM recommends a serum level of 20 ng/mL. For most adults, this requires an intake of 600–800 IU daily. However, the IOM also concluded that up to 4000 IU daily was likely safe for most adults. The target 25-(OH) vitamin D level for the patient at risk for GIOP should thus be 30 ng/mL (75 nmol/L). There is increasing literature emphasizing that excessive vitamin D may have deleterious effects, including nephrolithiasis and potential even deleterious effects on bone .
45.6.2
Guidelines for pharmacologic intervention and monitoring
Abnormalities in bone quality associated with initiation and, in particular, with use of higher dose glucocorticoids can occur even before deleterious effects on BMD are observed . For this reason, GIOP guidelines differ modestly in their use of BMD criteria to recommend treatment for new versus existing glucocorticoid users. In 2010 the American College of Rheumatology (ACR) Ad Hoc Guidelines Committee, for example, recommended pharmacologic therapy for new glucocorticoid users irrespective of BMD if glucocorticoid therapy is expected to continue for more than 3 months, although caution was advised in premenopausal women . For prevalent glucocorticoid users receiving prednisone doses of ≥5 mg/day who have a T score below −1.0, the ACR recommended pharmacologic intervention. For patients receiving doses of glucocorticoids >15 mg/day of prednisone, or for patients receiving lower doses (≥7.5 mg/day) but at high risk of future fracture, the UK and Dutch expert panels supported bisphosphonate use even in the absence of a bone mass measurement . This may be due in part to decreased availability of BMD testing in some populations. High-risk groups include postmenopausal women, men ≥70 years old, and individuals with a history of previous fragility fracture. Belgian guidelines recommend calcium and vitamin D alone for patients receiving 5–7.5 mg of prednisone-equivalents per day, as long as BMD is within the normal range . In situations in which bone mass measurement is impractical or unavailable, the guidelines suggested for the US Department of Veterans Affairs recommend empiric therapy with bisphosphonates when doses of prednisone ≥7.5 mg/day are prescribed for longer than 3 months .
The ACR updated its GIOP guideline in 2017 after a systematic review of the literature and use of contemporary evidence-based guidelines development techniques ( Figs. 45.10 and 45.11 ). Glucocorticoid-treated patients were characterized as low, moderate, or high fracture risk and further divided by age (<40 years old vs ≥40 years). For older adults, high risk was defined as a prior osteoporotic fracture, bone density T score ≤−2.5, or a FRAX adjusted for glucocorticoid dose score of ≤3% or ≤20% for 10-year risk of hip or major osteoporotic fracture (MOF), respectively. The authors also suggested, based on the studies of van Staa et al. that a very high dose (prednisolone equivalent of ≥30 mg daily) was also considered high risk. Moderate risk was considered FRAX scores of 1%–2.9% for hip and 10%–19% for MOF. Patients 40 or older with lower FRAX scores were considered low risk. Of note, the guideline authors recommended that for patients taking ≥7.5 mg of prednisone daily, the FRAX score should be increased by 20% (hip) and 15% (MOF). For those adults <40 years old, high risk was defined by the occurrence of an osteoporotic fracture, and moderate risk was defined as a Z score of <−3 or at least a 10% drop in hip or spine bone density in a year plus ≥7.5 mg of prednisone equivalent for ≥6 months. In addition to conservative management, such as adequate calcium and vitamin D, oral bisphosphonate treatment was recommended for older adults at moderate or high risk. While alternative treatment with intravenous bisphosphonates, teriparatide, and raloxifene could be considered, oral bisphosphonates were chosen as first line. Careful follow-up and reassessment of fracture risk were strongly recommended. Despite evidence from a 3-year head-to-head study that demonstrated fewer fractures in GIOP patients receiving teriparatide rather than alendronate, the authors of the GIOP guideline chose oral bisphosphonates as first line because of the expense and difficulty of daily injections with the anabolic drug, teriparatide.