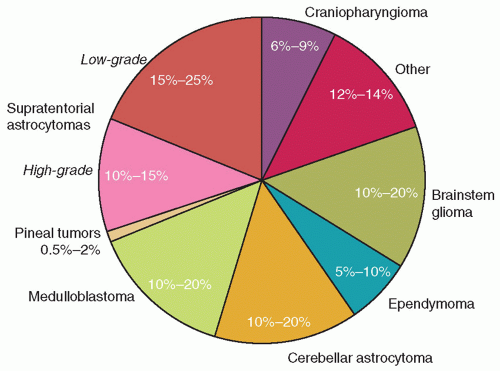
 Figure 26A.1 Approximate incidence of common CNS tumors in children. |
They may also develop medulloblastomas, primitive neuroectodermal tumors (PNETs), or choroid plexus tumors.5
TABLE 26A.1 Inherited Disorders Associated with Brain Tumors | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
at substantial risk of developing radiation-related secondary CNS cancers as a result of the location of the radiation field for their primary tumors.23 The Childhood Cancer Survivor Study (CCSS) has provided precise estimates for radiation therapy and the development of subsequent malignant neoplasms of the CNS.24
hematoxylin and eosin (H&E), other special stainings, immunohistochemistry, ultrastructural study, and cytogenetics can now be used to determine the cell types comprising the tumor with greater precision than was possible a decade ago. Use of monoclonal antibodies to identify specific antigens, such as cytoskeletal and membrane proteins, hormonal polypeptides, and neurotransmitter substances, has been especially instrumental in classifying, on routine light microscopy, tumors with unusual morphologic features that previously were relegated to the “unknown” category. Table 26A.3 contains a listing of the widely available markers that are used most commonly and their utility in the differential diagnosis of tumors arising in the CNS. Use of this phenotypic approach, coupled with increasing information of the cytogenetics and microarray analyses of CNS tumors, has forced changes in the historic classification schemes.
TABLE 26A.2 World Health Organization Classification of Tumors of the Nervous System (Neuroepithelial Tumors Only)31 | |
|---|---|
|
TABLE 26A.3 Common Markers for Diagnosis of CNS Tumors | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
not with accommodation. Convergence of gaze may evoke repetitive, bilateral, adducting nystagmus with retraction of the globes in the orbit. Cranial nerve IV palsy, with the affected eye deviated upward and laterally, may also occur. Affected children often compensate for the trochlear nerve palsy by tilting their heads toward the shoulder of the unaffected eye.
of a triad of long-tract signs, ataxia, and cranial neuropathies, particularly abducens palsy. The atypical, focal brainstem glioma presents with a longer prodrome, often without abducens palsy.
analysis is more difficult. With longer echo times, fewer peaks are captured, but the measurement precision is improved. In pediatric brain tumors, the three most important metabolic peaks (reading from right to left) are N-acetyl aspartate (NAA), 2.02 ppm; creatine-phosphocreatine (Cr/PCr), 3.02 ppm; and choline (Cho), 3.22 ppm (Fig. 26A.2).
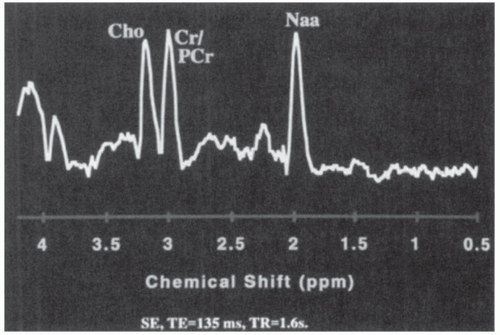 Figure 26A.2 Normal long-echo single-voxel spectrum from the cerebellum of an age-matched control for a patient with a posterior fossa brain tumor. Reading from right to left, note normal peaks of N-acetyl aspartate (NAA), creatine-phosphocreatine (CR/PCr), and choline (Cho). |
through the region of interest.66 Resulting data, reflected in maps of relative cerebral blood volume, provide some semiquantitative analysis of the blood flow to a particular region. Early work has suggested a positive correlation between relative cerebral blood volume and tumor grade.67 Perfusion imaging may be helpful in targeting a lesion for biopsy. Application of perfusion imaging may be particularly useful for the study of neovascularization and angiogenesis inhibition particularly using the T1WI technique. A 3D dynamic sequence is employed. Kinetic modeling of the dynamic signal changes can yield estimates of regional fractional blood volume and microvascular permeability (Kps), which is an indicator of BBB disruption and correlates with angiogenesis.68
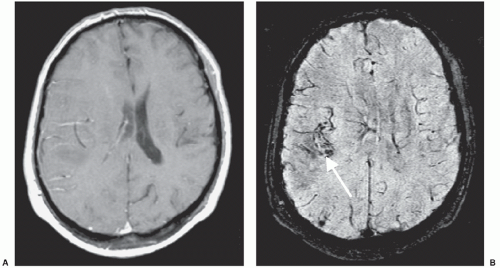 Figure 26A.3 A: T1-weighted MRI with contrast. B: Susceptibility-weighted image (SWI) showing hemorrhage in a glioma (arrow). (Courtesy of Mark Haacke, Ph.D., The Magnetic Resonance Imaging Institute for Biomedical Research, Detroit, Michigan.) |
18F-labeled agents such as fluorothymidine (FLT) for the assessment of cell proliferation and fluoro-DOPA and fluoromisonidazole for the assessment of protein synthesis and hypoxia within the tumor, respectively. Recent work in children with brainstem gliomas demonstrated that FDG-PET parameters correlated with survival.69
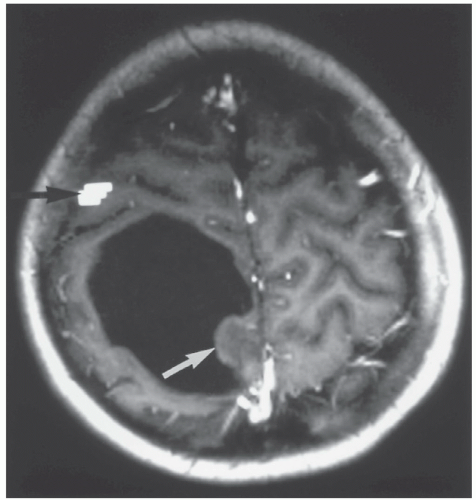 Figure 26A.4 Preoperative functional magnetic resonance imaging in an 11-year-old boy presenting with left-sided focal motor seizures, performed for surgical planning. The bright pixels (black arrow) represent the statistical parametric map produced by using a left-sided finger-thumb opposition paradigm. The bright pixels reflect the increase in oxygenated blood flowing away from the right-sided sensorimotor cortex. The white arrow indicates the solid tumor component within a cystic mass displacing the sensorimotor cortex anteriorly. Histopathology demonstrated a cystic dysplastic ganglioglioma that later was removed successfully via a posterior approach without damage to the child. |
for children with many types of pediatric brain tumors, particularly ependymomas,74 high-grade gliomas,75,76 medulloblastomas (see Chapter 26B), low-grade gliomas,77 and choroid plexus tumors.78 Accordingly, with the exceptions noted earlier, in which surgical resection is not indicated or in which stereotactic biopsy may be a preferable initial step, extensive resection is the goal for many types of pediatric brain tumors.
TABLE 26A.4 Management Schema for Pediatric Brain Tumorsa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
such as ependymomas and craniopharyngiomas, carry morbidity rates in excess of 40% with extensive resection.79 Although some studies have observed that morbidity is lower if operations are performed by neurosurgeons who do such operations frequently,80 other studies show that pediatric neurosurgeons are more likely to attempt extensive removals, on the basis of their recognition that this influences prognosis, and therefore may have overall rates of management of morbidity comparable with those of general neurosurgeons, albeit with a higher frequency of complete or nearly complete tumor resections.81
irradiation in children. With the introduction of sophisticated, 3D-image-guided radiation techniques capable of relative sparing of the normal brain structures, the focus of pediatric brain tumor trials has shifted to investigating broader indications for radiation as a key element in achieving disease control. Current studies are addressing ways to optimize the risk-to-benefit ratio of accurate, limited-volume radiation delivery, sometimes in the setting of reduced radiation dose as well.
and multi-beam plans do not significantly increase the risk of second malignancy and that the risk increases with dose.96
than 4 to 7 years.116 Deterioration in IQ is more prevalent in children following whole-brain or “focal” supratentorial radiation than after treatment confined to the posterior fossa.115
children with brain tumors not involving the hypothalamic-pituitary region and treated with either cranial or craniospinal irradiation. In patients receiving only cranial irradiation, the hypothalamic-pituitary region is estimated to receive a mean dose of 53.6 Gy (40 to 70 Gy).128
have been studied in adults154 and children155,156 with evidence of intriguing preliminary activity.
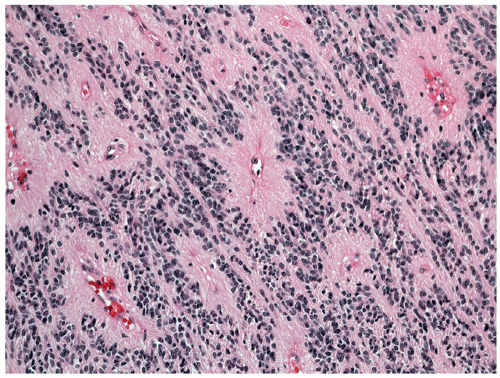 Figure 26A.5 Section from a fourth ventricular ependymoma displaying typical perivascular pseudorosettes (H&E, ×200). |
in the corresponding anatomic regions.170 More recent genome-scale and profiling analyses, including whole genome sequencing, have shown a relative paucity of focal copy number alterations and somatic mutations in ependymomas.171,172 but did identify a recurrent fusion (C11orf95–RELA) involving RELA, a key gene in the NF-κB signaling pathway, in approximately 70% of supratentorial ependymomas.171 These genomic data have provided evidence that ependymoma, like other pediatric CNS tumors, comprises a number of distinct molecular subtypes (C11orf95-RELA-positive and -negative supratentorial tumors, CPG island methylator phenotype [CIMP]-positive and -negative posterior fossa tumors, and spinal cord tumors).171,172,173 As a result of these studies, investigational treatment strategies targeting epigenetic mechanisms are of particular interest for children with ependymomas.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


