Gestational Vascular Complications
Benjamiun Brenner
Anat Aharon
Jean-Christophe Gris
PLACENTAL HEMOSTASIS IN NORMAL PREGNANCY AND GESTATIONAL VASCULAR COMPLICATIONS
The placenta is a highly vascularized organ receiving blood supply from fetal and maternal circulations. Maternal blood flows in the intervillous spaces, while fetal blood is confined in the intravillous blood vessels. This may result in potential hemostatic problems, mainly the risk of hemorrhage or placental vascular complications1 (FIGURE 117.1).
The placental villus is lined with trophoblast cells, specialized epithelial cells, that invade through the maternal endometrial stroma, migrate down the lumen of spiral arteries, replace the vascular endothelial lining, and become embedded in the arterial walls.2, 3, 4 The trophoblasts are involved in local hemostatic mechanisms as well as in maternal and placental crosstalk (FIGURE 117.1).
TISSUE FACTOR
During pregnancy, placenta and deciduas are putative sources of tissue factor (TF), which is localized on the cell membranes, can bind to factor VII, and initiate the coagulation cascade.5, 6 TF is also essential for embryogenesis, angiogenesis, invasion, and implantation.7, 8, 9 Normally, TF is not presented on cells that are in contact with blood circulation; however, the placenta is a unique organ—its surface is in direct contact with maternal circulation and trophoblast cells constitutively express high levels of TF antigen and activity3, 10 In addition, cytotrophoblast (CTB) differentiation and fusion to multinucleated cells—syncytiotrophoblasts (STB)—require initiation of apoptosis and exposure of negatively charged phospholipids on their membrane surface.11 Membrane “flip-flop” and exposure of negatively charged phospholipids are known to promote coagulation. Blockade of TF with a monoclonal antibody prevents activation of the coagulation pathway, restores placental blood flow, precludes placental oxidative stress, inhibits release of soluble vascular endothelial growth factor (VEGF) receptor 1 (sFlt-1), and consequently rescues pregnancy12 The procoagulant nature of placental trophoblast and decidual cells may reflect the physiologic need for immediate inhibition of hemorrhage in the placental intervillous spaces, protecting against placental abruption (PA) and meeting the hemostatic demands of labor and delivery.
PHYSIOLOGIC ANTICOAGULANTS
Despite the above-mentioned observations, coagulation does not normally occur across the entire villous surface.3 The high levels of TF are tightly regulated by a variety of physiologic anticoagulants—tissue factor pathway inhibitor (TFPI), endothelial protein C receptor (EPCR), thrombomodulin (TM), and annexin V—which act by a variety of mechanisms.
TFPI is highly expressed in placental tissues from the 10 weeks of pregnancy up to term.13, 14 TFPI is produced and pooled (50% to 80%) in the microvascular endothelial cells15 and in trophoblast cells.10, 13 In normal pregnancy, free TFPI antigen in plasma increased between first trimester and delivery but remained lower in nonpregnant controls.16
TFPI-2, (placental protein 5—PP5) is prominently expressed in human placenta17 and in isolated villous CTB cells,18 but its physiologic role in the placenta is currently unclear. Inactivation of the murine TF or TFPI genes results in embryonic lethality, indicating that both of these genes are required for embryonic development. Remarkably, the expression of low levels of TF in transgenic mice (1% of human TF) rescues TF-null embryos. However, low-TF mice (mTF(-/-)/hTF+) develop hemostatic defects in the uterus, placenta, heart, and lungs.19
TF-TFPI balance in human placenta may be disturbed, resulting in gestational vascular complications (GVCs). It has been found that placental trophoblasts obtained from GVC pregnancies have a disturbed balance between TF and TFPI. While some studies have demonstrated an increase in TF, others have revealed a decrease in TFPI, with underlying mechanisms remaining unclear.14, 20 Inflammatory cytokines, whose levels are occasionally elevated in GVC, contribute to this imbalance.10 Low molecular weight heparins (LMWHs) stimulate expression, synthesis, and release of TFPI in endothelial cells and may exert their effect in pregnant women at risk for GVC, by modulating local hemostasis at the placental STB surface.14
TM is a cell-surface receptor for thrombin that converts thrombin from procoagulant activity into the protein C (PC) anticoagulant pathway. Studies in mice demonstrated that abortion of TM-deficient embryos is caused by TF-initiated activation of the blood coagulation cascade at the fetomaternal interface. Activated coagulation factors induce cell death and growth inhibition of placental trophoblasts.21 The TM amount observed on vascular endothelium and STB rose significantly with growing gestational age, and the higher levels found at term microvilli were associated with an increase in TM ability to activate PC22 TM-mRNA expression increased in villous vessels of intrauterine growth restriction (IUGR) placentas23 and reduced in placentas of recurrent miscarriages compared to those of healthy pregnancies.24
Endothelial PC receptor enables PC activation by the thrombin-TM complex. In mice, EPCR is detected in the giant trophoblast cells at the fetomaternal boundary from E7.5. Disruption of the EPCR gene in mice causes placental thrombosis and early embryonic lethality.25 EPCR-/- embryos that had been removed
from extraembryonic membranes and tissues at day E7.5 and cultured in vitro developed beyond E10.5, suggesting a role of EPCR in the normal function of the placenta and/or at the maternal-embryonic interface.25
from extraembryonic membranes and tissues at day E7.5 and cultured in vitro developed beyond E10.5, suggesting a role of EPCR in the normal function of the placenta and/or at the maternal-embryonic interface.25
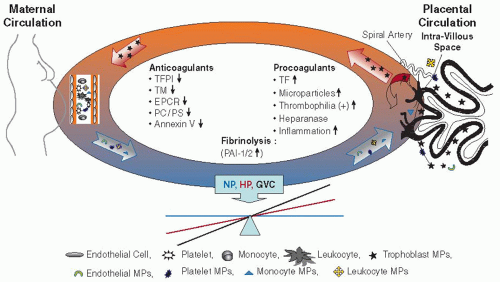 FIGURE 117.1 Placental-maternal crosstalk and hemostatic balance model. In pregnancy, the high levels of tissue factor (TF) on trophoblast cells and microparticles (MPs) are tightly regulated by a variety of physiologic anticoagulants: tissue factor pathway inhibitor (TFPI), endothelial protein C receptor (EPCR), thrombomodulin (TM), and anne×xin V, which act by a variety of mechanisms. In gestation vascular complications (GVCs), there are further increases in procoagulant properties, including maternal and placental MP levels, and further decreases in anticoagulant proteins, which perturb placental and maternal hemostatic balance. NP – non-pregnant HP – healthy pregnancy |
The common variations in EPCR gene (c.323-20T>C, c.717+16G>C, c.323-9_336 duplication, c.655A>G) are rare and are not a major cause of recurrent miscarriage. However, the EPCR A6936G polymorphism was found to be associated with the risk of unexplained fetal loss in Mediterranean European couples and paternal EPCR 219Gly variant was found to be a mild and limited risk factor for deep vein thrombosis during pregnancy26, 27, 28
Annexin V, an anticoagulant protein, is highly expressed on the apical surfaces of placental STBs. Binding of annexin V to the trophoblast procoagulant surfaces is crucial for the maintenance of blood flow through the placenta and consequently for fetal viability3, 29 Reduced annexin V expression has been demonstrated in some placentas from women with preeclampsia (PE)30 and fetal growth restriction.31 Anti-annexin V antibodies are associated with higher incidence of intrauterine fetal loss, PE, as well as arterial and venous thrombosis. In vivo study demonstrated that anti-annexin V antibody induced trophoblast apoptosis and significantly reduced trophoblast gonadotropin secretion.32
Heparanase. Heparan sulphate proteoglycans are abundant in the extracellular matrix (ECM) of placenta and deciduas. Heparanase cleaves heparan sulfate at specific sites, leading to a release of growth factors that may be involved in placentation and trophoblast invasion decidualization and remodeling of the maternal vasculature in humans and mice. Heparanase is expressed in normal and abnormal placenta, in small fetal vessels, and in a variety of trophoblast subpopulations with different invasive potentials.33, 34 A high level of heparanase was observed in isolated first-trimester trophoblasts; however, failure of blocking antibody to inhibit trophoblast invasion suggests another role for heparanase during pregnancy.35 Heparanase upregulates TF expression and interacts with TFPI on endothelial cells, resulting in increased cell-surface coagulation activity.36 The regulatory effect of heparanase on TFPI and TFPI-2 in trophoblasts suggests a potential involvement of heparanase in early miscarriages.37
Fibrinolysis. Fibrin accumulation, characterizing term placenta, can cause occlusion and infarcts in placental blood vessels and intravillous spaces and may result in placental insufficiency and pregnancy loss (PL). Fibrinolysis is a process ofclot degradation by plasmin that is converted from plasminogen by a tissue-type activator (tPA) or a urokinasetype plasminogen activator (uPA). Fibrinolysis is regulated by plasminogen activator inhibitor-1 and -2 (PAI-1, PAI-2) and by thrombin-activatable fibrinolysis inhibitor.38 Both decidual and extravillous trophoblast cells of human placentas are capable of producing tPA, uPA, PAI-1, and PAI-2 to a different extent. Variations in the distribution of tPA and PAI-1 may be related to fibrinolytic activity at early stages of placentation and to separation of placenta at term, while differences in the distribution of uPA and PAI-2 may be promoted by degradation of trophoblast cell-associated ECM at early stages of placentation.39 mRNA and protein levels of PAI-1 and PAI-2 were found to be higher in placental
tissue of preeclamptic women compared to healthy pregnancies. Exposure of trophoblast cell culture to tumor necrosis factor-α results in an increased expression and secretion of PAI-1, with no significant alteration in tPA.40, 41 The change in PAI-1and PAI-2 could explain the imbalance of local fibrinolysis at the placental trophoblast level in gestational complications.
tissue of preeclamptic women compared to healthy pregnancies. Exposure of trophoblast cell culture to tumor necrosis factor-α results in an increased expression and secretion of PAI-1, with no significant alteration in tPA.40, 41 The change in PAI-1and PAI-2 could explain the imbalance of local fibrinolysis at the placental trophoblast level in gestational complications.
MICROPARTICLES IN NORMAL PREGNANCY AND GESTATIONAL VASCULAR COMPLICATIONS
Microparticles (MPs), membrane vesicles, are found in blood circulation under normal physiologic conditions, and their levels increase in a variety of disease states. MPs are shed from cell membranes upon activation or apoptosis (FIGURE 117.2) and vary in size (0.1 to 1 µm) as well as in phospholipid and protein composition, reflecting those of their cell origin. MPs can induce cell signaling that may lead to invasion, angiogenesis, or apoptosis42, 43, 44, 45 and are involved in thrombosis, inflammation, and vascular dysfunction.46, 47 Therefore, MPs may have a potential significant role in the maternal placental crosstalk.
An overview of the studies measuring circulating levels of MP and their cell origin in normal and complicated pregnancy demonstrates high variation.48 Some of the trials showed an increase in MP subpopulations in women with PE compared to healthy pregnant females, including increase in the total number of MPs49 or in total negatively charged phospholipid-bearing MPs,50 elevation in platelet MPs50 and activated platelet MPs,51, 52 as well as increase in endothelial MPs53, 54 and white blood cell MPs.50, 55, 56 Other studies revealed no differences in the total number of MPs or negatively charged phospholipid-bearing MPs and platelet MPs between normal pregnant and preeclamptic women53, 55, 57 or decrease in the total negatively charged phospholipid-bearing MPs and platelet MPs51, 58 and activated platelet MPs59 in preeclamptic women compared to those with healthy pregnancies. The broad variation in the origin ofcell MPs observed in healthy pregnancy and PE may be related to the differences in the characteristics of GVC as well as to the diversity in study methods used for MP characterization.60
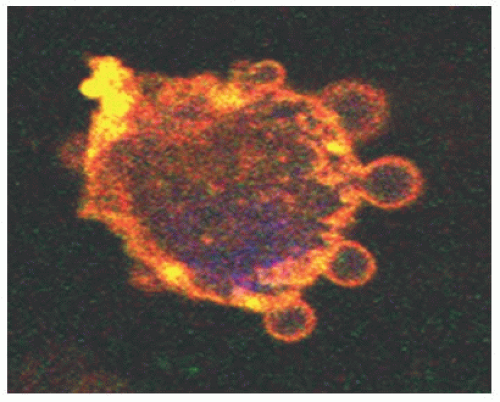 FIGURE 117.2 MP blebs from the membrane surface of umbilical vein endothelial cells (HUVECs) labeled with fluorescent-tagged antibody against CD31 and previously stimulated with monocyte MPs for 2 hours. This photograph was by confocal microscopy (60×). |
MICROPARTICLE THROMBOGENICITY
MPs play a dominant role in coagulation initiation and thrombus formation.47 MPs bearing TF and other coagulation factors may reflect the delicate hemostatic balance between maternal and placental cells (FIGURE 117.1).
MPs obtained from healthy pregnant women demonstrate high procoagulant activity compared to nonpregnant females. The procoagulant activity further increases in MPs of women with GVC without a change in the TF expression, but with reduction in TFPI.61 On the other hand, other trials demonstrated that MP-associated thrombin generation was similar in normal and preeclamptic pregnancies,62 the procoagulant activity generated by the total annexin V MPs was unchanged in pathologic pregnancies,58 and prothrombin fragments 1 + 2 and subsequent thrombin generation were nearly similar in women with recurrent spontaneous abortion and controls.63 The variance between the studies may be related, not only to the differences in the GVC characteristics and MP study methods, but also to the difference in coagulation assay sensitivity and diversity in the externalization patterns of negatively charged phospholipids and TF on MPs.64
TROPHOBLAST-DERIVED MICROPARTICLES
STB MPs are shed into the maternal circulation (FIGURE 117.1) and can be detected in normal pregnancies by the second trimester, while their numbers increase during the third trimester.65, 66 Excessive shedding of STB-derived MPs is a feature of preeclamptic women,65 mainly in early-onset PE, but not of normotensive IUGR.67 While normal pregnancy, labor, and placental separation have no effect on the shedding of STB MPs, PE is associated with an increase in STB MP shedding at full dilatation, compared to prelabor levels.68
Trophoblast differentiation is assumed to yield massive shedding of TF-bearing MPs that can be detected in the circulation of healthy pregnant women, and the percentage of total TF-bearing MPs significantly decreases in GVC. This represents an increase in the maternal source of TF-bearing MPs that potentially reflects the systemic nature of such pathologies.61
PE is characterized by a rise in inflammation markers. Higher amounts of circulating STB MPs in maternal blood might lead to endothelial dysfunction monocyte stimulation and excessive maternal inflammatory reaction.65, 66, 69, 70 Additionally, preeclamptic MPs have been shown to induce nuclear factor-KB activation and nitric oxide (NO) synthase expression and to enhance oxidative stress.71
In summary, the balance between the procoagulant TF and the physiologic anticoagulants is crucial for pregnancy and influences the placenta, embryo, and maternal crosstalk. The placental hemostatic balance has a major role in normal pregnancy, and the balance disruption may result in pregnancy complications. MPs affect physiologic processes (cell migration, invasion, and angiogenesis) and can promote pathologic states including thrombus formation, inflammation, and endothelial dysfunction. MPs participate in the placental and maternal crosstalk in normal pregnancies as well as in GVCs (FIGURE 117.1).
HEMOSTATIC CHANGES IN NORMAL PREGNANCY
Normal pregnancy is characterized by a marked increase in the procoagulant activity, manifested by an elevation of coagulation factors VII, X, and VIII; fibrinogen; and von Willebrand factor72 and an elevation in prothrombin fragments 1 + 2, and thrombin-antithrombin (AT) complexes,73 which is maximal around term. There is a profound decrease in physiologic anticoagulants, manifesting by a significant reduction in protein S (PS) activity74 and by acquired activated protein C resistance.75 The overall fibrinolytic activity is impaired during pregnancy but returns rapidly to normal following delivery,76 largely due to placenta-derived PAI type 2, which is present in substantial quantities during pregnancy77 Levels of D-dimer, a specific marker of fibrinolysis resulting from breakdown ofcross-linked fibrin polymer by plasmin, increase as pregnancy progresses.78
IMPLANTATION AND HEMOSTASIS
Pregnancy requires successful implantation of competent embryo into a receptive endometrium. Monthly fecundity rates in humans compared to other mammalian species are relatively low at approximately 20%.79 Of note, clinical pregnancy rates per embryo transfer for the year 2006 in Europe were around one-third.80 Current data suggest that thrombophilia can be associated with repeated implantation failure. One study reported significantly higher prevalence of thrombophilias in women with four or more failed IVF cycles when compared with spontaneous conceptions (OR 3.6, 95% CI 1.25 to 10.6) or women who conceived after their first cycle (OR 2.9, 95% CI 1.02 to 8.4).81 The mechanisms underlying these associations are less clear, partly due to the limited knowledge regarding the earliest stages of human implantation.
HEMOSTATIC CHANGES IN PREECLAMPSIA
Thrombocytopenia is a consistent finding in severe cases of PE closely correlating with activation of coagulation. A fall in platelet count in women with gestational hypertension may herald the onset of PE.82 The platelet membrane shows an abnormal lipid composition with a raised cholesterol to phospholipid ratio in PE.83 Assessment of platelet hyperactivity in whole blood may reveal elevated aggregation. Release of inflammatory mediators from activated platelets may trigger inflammatory reactions in endothelial cells. In theory, platelets are a potential regulator of trophoblast infiltration into maternal spiral arteries, thereby contributing to vascular remodeling.84 While at present the role of activated platelets in the pathophysiology of PE remains to be established, evidence of platelet activation preceding the onset of PE is emerging.85
Alterations in the hemostatic system leading to excessive uteroplacental thrombosis are thought to be an important feature at the first stage in the pathogenesis of PE, involving reduced placental perfusion due to failed vascular remodeling in early pregnancy within the placental bed.86 Coagulation factor levels including TF, factor VIII, fibrinogen, and von Willebrand factor are significantly higher in the peripheral circulation in PE compared with normal pregnancy. TF and free TFPI concentrations are significantly higher in maternal plasma of PE women compared with normal pregnancy87, 88 The TFPI to TF ratio is significantly lower in patients with PE than in normal pregnancy, and there is a higher maternal plasma concentration of PAI-1 in PE compared to a normal pregnancy. Of interest, increased PAI-1 levels are detected in patients who show early evidence of placental dysfunction on uterine artery Doppler studies, while the low PAI-2 levels in PE may reflect placental dysfunction.
EPIDEMIOLOGY AND PATHOGENESIS OF PREGNANCY COMPLICATIONS
Unexplained PL and GVC, including PA, PE, and small-for-gestational age (SGA) pregnancies, are heterogeneous gestational pathologies associated with devastating maternal and fetal complications, variably affecting 5% to 15% of all pregnancies.
Up to 9% to 13% of women experience one clinically recognized PL, 5% two or more PLs, and 1% to 2% three or more losses.89, 90, 91, 92, 93 Up to 50% of recurrent PLs remain unexplained, with unclear initial categorization94 (Table 117.1). Early measurements of the fetus crown-rump length (CRL) and detection of fetal heart beats led to the definition of gestational age based on ultrasound (US)95 assessment (“fetus” meaning fetal heart activity and/or a CRL > 10 mm), with four PL categories being recognized95 (Table 117.2). Women with idiopathic PL tend to have recurrences at the same gestational age period,96 with a likelihood of PL causes to be gestational age specific. Placental bed biopsies from karyotyped, late97 and early98 sporadic miscarriages suggest that only late miscarriages are associated with impaired trophoblast invasion and inadequate transformation of spiral arteries, similar to PE. Most recurrent PLs are nonfetal cases resulting from random repetition of aneuploidic pregnancies; normal embryonic karyotypes increase with the number of previous PLs ( = 2: <50%; >6: 75%)99 and predict a subsequent PL.100 First-trimester idiopathic recurrent PL occurs in 22%, 2%, and 0.6% before 6, 8, and 10 gestational weeks, respectively with only 3% following the detection of fetal heart activity101 Recurrent PL means at least three losses, usually of unknown cytogenetics: two euploid PLs identify couples at higher recurrence risk, likely to bear a causal noncytogenetic factor.102
PE complicates 4% to 10% of nulliparous pregnancies and is twofold more frequent in primigravidas.103 The recurrence risk is around 15% after one previous PE, increasing to 30% after two consecutive PEs.104 A severe recurrent early-onset type, with delivery before 34 weeks, affected by chronic genetic/environmental factors, opposes a milder sporadic form affected by transient factors (two-third of cases).104 Severe PE subtypes are associated with various recurrence risks. Eclampsia generates a
mean 22% PE recurrence risk and a 2% eclampsia risk. In primigravidas, early-onset PE before 34 weeks generates a mean 25% PE risk and a 5% early-onset PE risk during the second pregnancy. Moreover, a second-trimester PE generates a 66% PE risk on the next pregnancy. Likewise, HELLP syndrome (hemolysis, elevated liver enzymes and low platelet counts) generates a mean 43% PE and a 27% HELLP syndrome recurrence risk. While smoking during pregnancy is associated with spontaneous abortion, stillbirth, preterm labor, fetal growth restriction, and PA, the incidence of PE is reduced in smokers.105 Abnormal placentation and an imbalance in angiogenic factors are essential for the development of the PE syndrome,106 with high levels of trophoblast-derived circulating antiangiogenic factors mediating maternal endothelial dysfunction, soluble fms-like tyrosine kinase-1, and soluble endoglin.
mean 22% PE recurrence risk and a 2% eclampsia risk. In primigravidas, early-onset PE before 34 weeks generates a mean 25% PE risk and a 5% early-onset PE risk during the second pregnancy. Moreover, a second-trimester PE generates a 66% PE risk on the next pregnancy. Likewise, HELLP syndrome (hemolysis, elevated liver enzymes and low platelet counts) generates a mean 43% PE and a 27% HELLP syndrome recurrence risk. While smoking during pregnancy is associated with spontaneous abortion, stillbirth, preterm labor, fetal growth restriction, and PA, the incidence of PE is reduced in smokers.105 Abnormal placentation and an imbalance in angiogenic factors are essential for the development of the PE syndrome,106 with high levels of trophoblast-derived circulating antiangiogenic factors mediating maternal endothelial dysfunction, soluble fms-like tyrosine kinase-1, and soluble endoglin.
Table 117.1 Classical definitions of PL types | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Inherited Thrombocytopenias
Inherited Thrombocytopenias
 Unusual Sites of Arterial Occlusion
Unusual Sites of Arterial Occlusion
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


