Genomics, Prognosis, and Therapeutic Interventions
Genomics, Prognosis, and Therapeutic Interventions
Lisa A. Carey
Maggie Chon U Cheang
Charles M. Perou
CURRENT OVERVIEW
The rapid advancement of high-information content technologies has resulted in the completion of numerous molecular profiling studies of breast cancers. Global gene expression profiling, massively parallel sequencing (MPS), array-comparative genomic hybridization (aCGH), and reverse phase protein arrays (RPPA) have allowed scientists to profile RNA, DNA, and proteins in hundreds of human breast tumor tissues in a speedy fashion. To fully decipher the mountains of data generated from these ‘omics’ approaches, and to translate the findings into the clinical setting is challenging, but, undoubtedly, is also one of the highest research priorities for the next few years. In this chapter, we will review how genomics have informed our understanding of the heterogeneity of breast cancer and how it is currently being used for prognostication and therapeutic decision-making. The promise of gene expression patterns, when possibly coupled with somatic mutational profiles, is the near future when we will be able to use the detailed tumor-specific, and patient-specific, information as a means to personalize therapy for breast cancer patients.
Breast cancer is a known heterogeneous disease comprised of a growing number of recognized biologic subtypes. Clinicians and researchers have noted the variations in risk factors, response to therapy, and clinical behavior according to hormone receptor status (i.e., Estrogen Receptors [ER] and Progesterone Receptors [PR]) for several decades. More recent data has also implicated HER-positive breast cancers as possessing unique characteristics, such as responsiveness to anthracyclines (1). Traditional single marker approaches to biomarker identification is limited by the fact that seldom is one gene/protein responsible for the entire action of a cellular pathway. Even more importantly, single marker studies do not address the important relationships among and within different pathways, which are increasingly becoming needed to predict tumor behavior and response to therapy.
As mentioned earlier, MPS is a new and powerful tool for the study of human cancers. The first commercially available MPS platform was the 454 technology by Roche Applied Sciences (2). As of today, other platforms available in the market include the HiSeq and MiSeq Systems by Illumina, Ion Torrent PGM and Proton by Life Technologies, and PacBio RS by Pacific Biosciences. Each technology uses a proprietary approach to sequence molecules of DNA; however, all result in the generation of tens of thousands, to even tens of millions, of sequence ‘reads,’ which are then used to reconstruct the genomic DNA sequence (or mRNA sequence) of a gene or genome. Although the clinical integration of targeted sequencing assays may still be a few years away, in 2012, eight published landmark studies all applied MPS and/or other DNA-based ‘omic’ technologies to create a comprehensive catalog of somatic mutational events that are driving breast cancer pathogenesis (3, 4, 5, 6, 7, 8, 9 and 10). This new MPS data, and the older gene expression array data, provide the genetic framework for personalized medicine as follows.
GENE ARRAYS
DNA microarrays have been used as a means to better understand tumor biology and to predict outcomes and response to therapy. Gene expression microarrays measure the level of expression of a particular gene by quantitatively determining the level of mRNA transcripts, which can be initially compared to a reference sample (as in the case of two-color arrays like those produced by Agilent), or directly compared to other tumors or normal tissues (as in the case of one-color arrays like those produced by Affymetrix). The arrays themselves currently include essentially all human genes that are encompassed in thousands to millions of features/probes that are either oligonucleotides, or PCR amplified cDNA inserts. These sequences represent known, unknown but validated, and hypothetical genes, and all approximately 37,000 genes in the human genome can be included (˜25,000 protein coding genes, ˜12,000 non-coding RNAs). A tumor is processed for RNA, which is used to generate either complementary DNA or RNA, labeled with a fluorescent probe. These fluorescent probes are then either directly applied to a gene array alone (one-color arrays), or combined with a second fluorescently labeled reference sample and then both are applied to an array (two-color array, in which, by convention, the color ‘red’ is used for the sample of interest and the color ‘green’ for the common reference sample). The remainder of the assay is basically a Southern blot with nucleic acid hybridization reactions occurring and binding, and with the intensity(s) of the nucleic acids that hybridize to the individual gene probes reflecting the relative amounts of tumor mRNA. In the case of a two-color microarray the ‘green’ signal predominance reflects low expression and ‘red’ predominance reflects high expression of that gene in the tumor relative to the reference (Fig. 29-1). Thus, in a twocolor array it is not the value of the tumor versus the reference that is of greatest interest, but the ratio of tumor/reference for each gene that is used as a quantitative measure of that gene. This value is then used to compare tumor to tumor, and tumor to normal; in this way, once a two-color microarray gives a tumor/reference value, it is used nearly identically to the onecolor microarray absolute intensity values, and thus, once the user gets past these initial different data processing steps, all downstream analysis steps are similar.
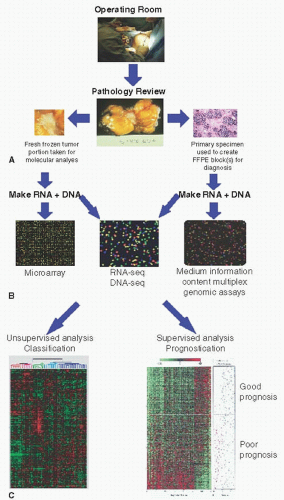
FIGURE 29-1 Overview of gene expression analysis of human tumors. (A) Tumors are collected in the operating room and often split into multiple aliquots. (B) Nucleic acids (i.e., RNA and DNA) are isolated and can be assayed on a variety of technologies. (C) Unsupervised and supervised analyses are performed to identify tumor subtypes and/or to develop predictors of outcomes or response to therapy.
MOLECULAR PROFILING OF BREAST CANCERS BY GENE EXPRESSION ARRAYS
Most breast cancer molecular profiling studies have focused upon gene/mRNA expression. Multiple approaches to analyzing gene expression microarrays exist; however, it is self-evident that the answer one gets from genomic analysis depends upon the question one asks. For example, unsupervised analyses (which use no external guide) can identify whether there are molecularly identifiable tumor subtypes, also called “class discovery,” while supervised analyses (analyzed with a particular clinical endpoint in mind) can identify if there are genes correlated with relapsed patients versus those that did not relapse (prognostic profiles), or related to patients who responded to a particular therapy versus those that did not (predictive profiles), which are also called “class comparisons” (11, 12). It is tempting to assume that the individual genes that are identified using these methods may themselves be causal in creating the subtype, or the clinical characteristic, and sometimes they are; however, in truth, the identified genes themselves may be mere proxies for genetic events or pathway activation and may not themselves be the cause. A third category of analysis, called “class prediction,” first uses a prespecified gene list (typically coming from a supervised analysis) and a given sample set (and classification rule) that is then used to assign a new individual tumor to a particular category, such as a genomic-based class; all genomic tests offered in the clinic should have reached the class prediction stage.
The molecular profiling of breast cancer has provided important information for three major questions: (1) Are there subtypes of breast cancer based on biological differences? (2) Are there gene expression profiles that can distinguish poor from good prognosis patients, thus allowing us to make better-informed decisions regarding adjuvant therapy? (3) Are there gene expression profiles that can predict which tumor will respond to a specific therapy? These questions, and others, will be addressed in the following sections.
BREAST CANCER INTRINSIC SUBTYPES
In 2000, Perou and colleagues used a semi-supervised approach to identify naturally occurring breast cancer subtypes in a population of 40 patients with locally advanced disease treated with neoadjuvant chemotherapy (13). They identified 496 genes termed the “intrinsic gene set” that showed little variance within repeated tumor sample, but high variance across different tumors, and then used this gene set for potential subtype discovery. Among these breast cancers, they found that the patterns of expression of these genes segregated the tumors into four subtypes, and in Sorlie et al. (2001), they identified a fifth possible subtype (14). The five ‘intrinsic’ subtypes are so-called because the gene list that defines them reflects intrinsic properties of breast cancers rather than being contributed by other cell types or augmented by drugs. These subtypes have been consistently identified in independent datasets using multiple different technologies (15, 16, 17, 18, 19, 20 and 21), are conserved across ethnic groups, and are present in preneoplasia (21, 22). Reassuringly, the intrinsic subtypes are segregated by expression of hormone receptors and the genes they regulate (and actually include ER, PR, and HER2), supporting earlier epidemiologic and biomarker studies suggesting that ER positive and ER negative breast cancer are different. At least two hormone receptor positive subtypes were identified and are called “Luminal A” and “Luminal B.” Conversely, there are several subtypes characterized by low expression of hormone receptors, one of which is called the “HER2-enriched” subtype (HER2-E) and another called the “Basallike” subtype (Fig. 29-2A). The fifth subtype, the normal-like, is less clearly a subtype rather than a likely technical artifact possibly caused by too much normal contaminating tissue. A new possible subtype, named Claudin-low, has been recently identified, which is characterized by low to absent expression of cell adhesion genes including Claudin 3, 4, 7, and E-cadherin (23). Although the intrinsic subtypes were identified without any knowledge of outcomes, these subtypes have strong prognostic implications (Fig. 29-2B); in particular, patients with Basal-like, Claudin-low, HER2-E, or Luminal B tumors demonstrate a significantly worse outcome compared to patients with Luminal A tumors in datasets from patients treated with no systemic adjuvant therapy, and in patient sets treated more heterogeneously including adjuvant and neoadjuvant chemotherapy (15, 16, 17, 18 and 19, 24, 25).
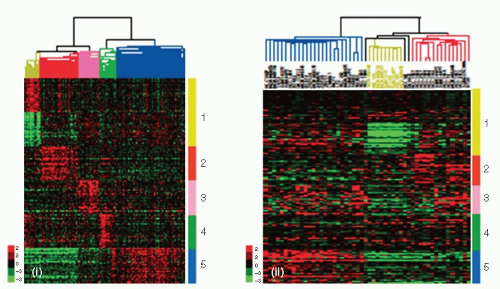
FIGURE 29-2A (A) Gene clusters that characterize each primary human tumor subtype are shown in the human (i) and cell line (ii) gene expression data sets. In both data sets, array trees have been derived by unsupervised hierarchical clustering using the 1,906 intrinsic genes as described in Parker et al. (29). (i) The top 50 upregulated genes associated with each molecular subtype, including the top 50 downregulated genes in Claudinlow tumors, are shown in the UNC337 database that included 320 breast carcinomas and 17 normal tissues. Top genes were selected after performing a two-class Significant Analysis of Microarray (SAM) (false discovery rate = 0%) between each molecular subtype versus others. Luminal A and B subtypes were combined into the luminal subtype. In the tree, the yellow node denotes the Claudin-low tumors. (ii) Gene clusters characteristic of each tumor molecular sub-type are shown in 52 breast cancer cell lines. Missing genes have been omitted. In the tree, the yellow node denotes the most highly correlated cell lines that best resemble the Claudin-low subtype. 1 (yellow), Claudin-low gene cluster of upregulated and downregulated genes; 2 (red), basal-like gene cluster; 3 (pink), HER2-enriched gene cluster; 4 (green), normal breast-like gene cluster; 5 (blue), luminal gene cluster. (Reproduced with permission from Prat A, Parker JS, Karginova O, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Research 2010;12:R68.)
A critical aspect of biomarker biology is validation and the intrinsic subtypes have been validated through multiple common findings including similar distributions on many independent datasets, and similar overall risks/prognoses as well (14, 17, 19, 26). Because the clustering methodology for the initial identification of the intrinsic subtypes is suboptimal for everyday clinical classifications, the development of a robust subtyping method for individual patient samples has been an area of active research. One of the promising approaches for reproducible subtype classifications is based upon identifying a subtypes mean expression profile, called a centroid (17, 18, 27). Hu and colleagues developed the Single Sample Predictor (SSP) tool to serve as a first generation, unchanging classifier for individual patient samples; the SSP compares the gene expression profile of an unknown sample to a prototypical profile of each intrinsic subtype and classifies the unknown sample according to the profile it most closely matches. Thus, one sample or hundreds of samples can be objectively classified in a reproducible fashion.
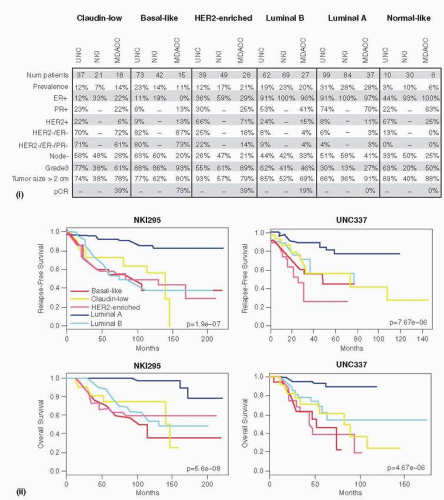
FIGURE 29-2B Clinical and pathological characteristics and prognosis of all intrinsic subtypes, including Claudin-low tumors, across three independent breast cancer data sets. (i) Percentages of the different clinical-pathological characteristics in the UNC337 data set and two publicly available data sets (NKI295 and MDACC133 [117]). ER/PR/HER2 scores of the UNC337 database were based on clinically validated methods. (ii) Survival data of the different molecular subtypes are shown for the UNC337 database and NKI295 (26, 87). Normal breast-like samples have been removed from this analysis. The UNC337 set represents a heterogeneously treated group of patients treated in accord with the biomarker status, whereas NKI295 is predominantly a local therapy only cohort. (Reproduced with permission from Prat A, Parker JS, Karginova O, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Research 2010;12:R68.)
Fresh frozen (FF) tumor samples are usually preferred for microarray experiments, therefore limiting the practicability of these approaches for correlative sciences on clinical trials when often only formalin-fixed paraffin embedded (FFPE) tissues are available. However, Mullins and colleagues demonstrated that there was 94% concordance (33 of 35 matched FF-FFPE pairs) when comparing the subtype assignments by centroid-based algorithm from FF tissue by microarray versus FFPE tissue assayed by real-time quantitative reverse transcription-PCR (qRT-PCR) (28). In 2009, Parker and colleagues developed an improved, opensource intrinsic subtype classifier based on a minimal list of 50 genes, commonly known as the PAM50 (29). In this study, 122 matched frozen-FFPE tumor pairs were subjected to both microarray and qRT-PCR analysis, and a centroidbased predictor was developed that used 50 genes, and which could use either FF RNA (microarray) or FFPE RNA (qRT-PCR). This 50-gene subtype predictor provided significant prognostic value independent of the standard clinical parameters in a test set of 761 patients receiving no systemic therapy, and significantly predicted the pathologic complete response (pCR) in a test of 133 patients treated with neoadjuvant taxane and anthracycline regimens (29).
In the same study, the authors also developed several risk models based upon a Cox Modeling approach, which uses the genomic-determined subtype data and a standard clinical variable (tumor size). A Risk of Relapse (ROR) score could be assigned to each test case (that is, a patient sample) using a Cox model based upon a tumors “distance” to the subtype centroids alone (ROR-S), or using subtype correlation along with tumor size (ROR-T) (29). The most current standard for molecular intrinsic subtyping is the PAM50 50-gene test, which includes the above subtypes except the Claudin-low, and is undergoing extensive clinical validation (30, 31, 32, 33 and 34).
Other approaches to defining the intrinsic subtypes have been attempted using non-microarray-based methods, most often using immunohistochemistry (IHC) (35, 36). However, the accuracy using an IHC-based approach is not as great as the multi-gene expression assays, mostly because IHC assessments can be subjective, and suffer from inter-observer and inter-laboratory variations. Despite the limitations of IHC, performing tumor subtyping via IHC is still valuable and has been adopted as a means of classification by the St. Gallen’s consensus conference (37); specifically, the indication is that Luminal A patients as defined by this IHC-based definition (i.e., ER+ and/or PR+, HER2-normal, Ki-67 less than 14%), may not be recommended to receive adjuvant chemotherapy given their overall general good prognosis.
Luminal Intrinsic Subtypes
The most common subtypes of breast cancer are the Luminal subtypes, so-called because they have a gene expression pattern reminiscent of the luminal epithelial component of the normal breast (13). These tumors are characterized by expression of the estrogen receptor (ER), progesterone receptor (PR), and genes associated with ER activation such as LIV1, TFF1, and Cyclin D1, as well as expression of luminal cytokeratins 8 and 18 (13, 20, 38). Within the luminal tumors family there are at least two subtypes, Luminal A and Luminal B, and there are many relevant differences between these two groups. For example, Luminal A tumors generally have high expression of GATA3, ER-regulated genes including PR, low expression HER2 and of the HER2 amplicon expression cluster, and low expression of proliferation-associated genes including Ki-67 (19, 25). Conversely, Luminal B tumors tend to be highly proliferative, are sometimes HER2+, and show lower expression of PR (36, 39) and of other ER-regulated genes (36, 39).
When compared to other breast cancer subtypes, Luminal tumors have a low frequency (fewer than 20%) of TP53 mutations (20, 25), with a rate of 12% in Luminal A and a higher frequency of 29% in Luminal B tumors (4), with the presence of mutant TP53 being strongly associated with endocrine therapy resistance (6). Interestingly, ˜30% of Luminal B tumors also have amplification of the TP53 antagonist MDM2 (4), thus suggesting that inhibitors for MDM2-p53 interaction might be a potential treatment approach for a subset of the aggressive Luminal B tumors. In addition, The Cancer Genome Atlas (TCGA) data also reported that phosphatidylinositol-3-kinase (PIK3CA) mutations are the most common Significantly Mutated Gene (SMG) in Luminal breast cancers (˜40%), hence another potential therapeutic target. In addition, the site of PIK3CA mutation may be subtype-specific where, for example, almost all the “hotspot” E545K mutations occurred in Luminal A subtype (25/27), whereas the other common hotspot (i.e., H1047R) occurred in all of the subtypes (4). Whether adjuvant studies focusing on PIK3CA mutations should stratify by mutation type and intrinsic subtype is debatable, but should be kept in mind as the high correlation between subtype and mutation type is likely an important biological feature of these Luminal A cancers.
In population-based studies that classified tumors using IHC, Luminal A breast cancer is the most common, representing approximately 40% to 50% of tumors while Luminal B comprises approximately 10% to 15% (16, 35, 36, 40, 41). Expression array-based profiling studies suggest that Luminal A comprises approximately 30% to 40% and Luminal B approximately 20% of breast cancers (17, 42), with Luminal A breast cancers consistently showing a better prognosis than Luminal B (19, 20, 25, 43). Although life history risk factors for all of the subtypes remain complex, it is increasingly clear that most traditional risk factors are primarily risk factors for Luminal breast cancers (44). In addition, the population-based studies also show that premenopausal women, and African-American (AA) women, tend to develop fewer of the good-prognosis Luminal tumors and more of the poorprognosis Basal-like tumors (described further later), which may contribute to the worse mortality outcomes experienced by groups (i.e., young women, and/or AA women) (16, 41). While clinical gene expression-based assays to identify Luminal A and B are not yet formally available, the OncotypeDx Recurrence Score™ (RS) assay includes many genes (HER2, GRB7, ER, SCUBE2, Bcl2, Ki-67, Survivin, MYBL2, and Cyclin B1) that are also used to define Luminal A vs. Luminal B tumors. To more directly compare intrinsic subtyping to the Recurrence Score, Fan et al. ran a research version of both assays on a single data set of patients and showed that 50% of 123 Luminal A tumors had low RS (associated with good outcome) whereas only 2% of Luminal B tumors had low RS (with almost all Luminal B being called RS high) (42) (Table 29-1). On the other hand, Kelly et al. compared the risk assignments between OncotypeDx RS and the research version of intrinsic subtypes by PAM50 qPCR classifier on 151 ER positive stage I-II tumors (45). Seventy percent of Luminal As had low RS and 90% of the high RS tumors were Luminal B. These concordant findings from two different genomic assays validate the genomics approach in general, and highlight the biomarker powers of multi-gene expression assays.
TABLE 29-1 Prognostic Profile by Intrinsic Subtype
Recurrence Score
70 Gene Profile
Wound Response
Intrinsic Subtype
No. of Patients
Classification
No. of Patients
Classification
No. of Patients
Classification
No. of Patients
Low
0 (0%)
Good
0 (0%)
Quiescent
3 (6%)
Basal-like
53
Intermediate
High
Low
0 (0%)
53 (100%)
62 (50%)
Poor
Good
53 (100%)
87 (71%)
Activated
Quiescent
50 (94%)
45 (37%)
Luminal A
123
Intermediate
High
Low
25 (20%)
36 (29%)
1 (2%)
Poor
Good
36 (29%)
9 (16%)
Activated
Quiescent
78 (63%)
4 (7%)
Luminal B
55
Intermediate
High
Low
4 (7%)
50 (91%)
0 (0%)
Poor
Good
46 (84%)
3 (9%)
Activated
Quiescent
51 (93%)
0 (0%)
HER2+/ER-
35
Intermediate
High
Low
0 (0%)
35 (100%)
7 (24%)
Poor
Good
32 (91%)
16 (55%)
Activated
Quiescent
35 (100%)
15 (52%)
Normal-like
29
Intermediate
High
4 (14%)
18 (62%)
Poor
13 (45%)
Activated
14 (48%)
Adapted from Fan C, Oh DS, Wessels L, et al. Concordance among gene-expression-based predictors for breast cancer. N Engl J Med 2006;355:560-569.
HER2-Enriched Subtype
The hormone receptor-negative subtypes are comprised of the HER2-Enriched (HER2-E) and Basal-like subtypes, although it should be noted that not all HER2-E, and not all Basal-like tumors, are ER/PR negative. The HER2-E subtype has elevated expression of HER2 and many other genes that reside near HER2 in the genome including GRB7 (see section on Recurrence Score) because of HER2 region genomic DNA amplification. These tumors also show low expression of the luminal, hormone receptor-related gene cluster, and low expression of the Basal-like cluster. However, it is imperative to note that many, but not all, clinically defined HER2-positive breast cancers fall into the HER2-enriched category; for example, ˜55% of the clinically defined HER2-positive breast cancers (30, 46) were ER positive tumors that were classified as luminal subtypes, thus, there exists at least two types of clinically HER2-amplified tumors (i.e., HER2-E and Luminal/HER2+).
Another important feature of tumors in the HER2-Enriched subtype is high expression of the proliferation cluster and, befitting this expression pattern, 75% are high grade tumors and over 70% have p53 mutations (4). This subtype is uncommon, comprising only 5% to 10% of all breast cancers in population-based studies (16). In the era before HER2-targeted therapy, the HER2+/ER- subtype carried a poor prognosis (19, 20, 25, 43, 47). Given that there is no apparent interaction between the benefit of HER2-targeted therapy such as trastuzumab and hormone receptor status, it is reasonable to presume that the HER2-E subtype has benefited from the HER2-targeting revolution to the same degree as a HER2-positive Luminal breast cancer (48). The risk factor profile for the HER2-Enriched subtype mirrors the other Luminal tumor subtypes (49), and there is no apparent interaction with race or age (16, 41). Aside from HER2 amplification and TP53 mutation, other frequent somatic mutation alterations include PIK3CA (39%) and another PI3K-pathway component (PIK3R1), but with a much lower frequency of 4% (4).
In the clinic, it is known that approximately 50% of patients with clinically HER2+ tumors respond to HER2-targeted therapies like trastuzumab. The TCGA data may provide a rationale for this as one subgroup of clinical HER2+ disease showed high levels of epidermal growth factor receptor (EGFR) and HER2 protein phosphorylation, and this subgroup was largely coincident with the HER2-Enriched subtype (4), whereas the other clinical HER2+ tumors showed the luminal phenotype and lower levels of phosphorylated EGFR and HER2. Whether the HER2-E subtype could be a biomarker for trastuzumab and/or lapatinib sensitivity, and/or HER2 and EGFR protein phosphorylation, could serve as predictive biomarkers, is yet to be determined.
Basal-Like Subtype
The Basal-like subtype is characterized by low expression of the hormone receptors and luminal subtype-related genes, and low expression of HER2 (and lack of gene amplification), thus, most of these tumors are of the so-called “triple negative” classification; however, not all Triple Negative Breast Cancers (TNBC) are Basal-like, and not all Basal-like cancers are TNBC (50) (Fig. 29-3). Other expression features of Basallike cancers include high expression of the proliferation signature, and high expression of a unique cluster of genes called the basal cluster. The basal gene cluster includes typical basal epithelial cytokeratins (CK) such as CK5, 6, 14, and 17, the epidermal growth factor receptor, c-Kit, Vimentin, P-Cadherin, and αB-crystallin. Massive parallel sequencing studies have shown that Basal-like tumors are molecularly distinct from Luminal tumors, and are more similar with tumors arising in the basal layer of epidermis including squamous carcinoma of the lung (51), and epithelial ovarian cancer (4). Although the PIK-pathway appears activated within Basal-like tumors, unlike Luminal subtypes, there is a low frequency of PIK3CA mutations (9%) (4), but higher frequency of PTEN (35%) and INPP4B (30%) loss (i.e., the deletion/mutation of negative regulators for PIK-pathway) (4). Similar to HER2-E subtype, the presence of TP53 somatic mutations (85%) is extremely frequent within Basal-like tumors, which is another property they share with serous ovarian cancers (95% TP53 mutant) (52).
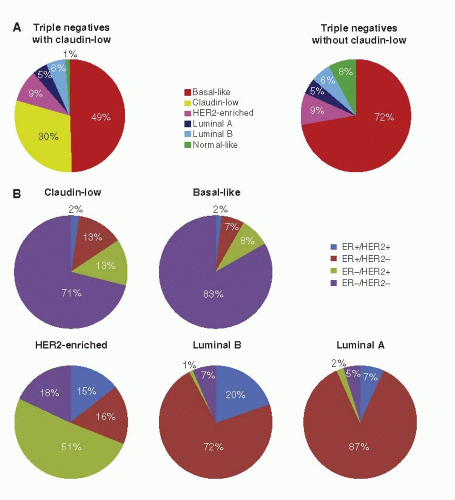
FIGURE 29-3 Distribution of clinicalpathological categories relative to the intrinsic subtypes. (A) Intrinsic subtype distribution within the triple-negative tumor category shown with and without Claudin-low tumors. (B) Distribution of ER+/HER2+, ER+/HER2+, ER-/HER2- clinical groups in the Claudin-low, Basal-like, HER2-enriched, Luminal B, and Luminal A within each subtype. (From Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Molec Oncol 2011;5:5-23.)
Several risk factors for developing Basal-like subtype tumors have been identified with one of the most intriguing being the link between the Basal-like subtype and BRCA1 mutation carriers (19, 53, 54 and 55). Women who carry a deleterious mutation in BRCA1 are at > 50% risk of developing breast cancer, and, when they do, over 80% of the time it is Basal-like. However, while BRCA1 mutation carriers usually develop Basal-like breast cancer, most Basal-like breast cancers are sporadic and the BRCA1 gene and protein appear intact in these tumors. A commonly held, but unproven, assumption is that the BRCA1 pathway is somehow deranged in sporadic Basal-like breast cancer, which, if true, could have important therapeutic implications. From the TCGA data, there is a combined frequency of 20% for BRCA1 and BRCA2 mutations (both germline and somatic) in the Basallike subtype (4). The BRCA1/2 pathways, which include a number of other genes such as FANC genes, are involved in homologous recombination mediated DNA repair, which is a high fidelity DNA repair pathway. When the homologous recombination pathway is lost or dysfunctional, DNA repair occurs by the more error-prone methods that involve poly (ADP-ribose) polymerase (PARP), which can be inhibited by a novel class of drugs that are being tested in clinical trials (56, 57). Loss of normal DNA repair is also implicated in sensitivity to chemotherapy, particularly to DNA-damaging agents such as platinum drugs (58), although recent studies suggest that Basal-like breast cancers may have a general sensitivity to chemotherapy (30, 47, 59). Gathering the commalities between Basal-like subtype and high-grade serous ovarian cancers together, one might predict that platinumbased chemotherapy might be a potential therapeutic option for Basal-like tumors.
Only gold members can continue reading. Log In or Register to continue
 Breast Cancer Screening
Breast Cancer Screening
 Ductal Carcinoma In Situ and Other Intraductal Lesions: Pathology, Immunohistochemistry, and Molecular Alterations
Ductal Carcinoma In Situ and Other Intraductal Lesions: Pathology, Immunohistochemistry, and Molecular Alterations
 Adjuvant Systemic Therapy: Endocrine Therapy
Adjuvant Systemic Therapy: Endocrine Therapy
 Preoperative Endocrine Therapy for Operable Breast Cancer
Preoperative Endocrine Therapy for Operable Breast Cancer



