Genomic Events in Breast Cancer Progression
Rodrigo Goncalves
Burcu Cakar
Ron Bose
Cynthia X. Ma
Matthew J. Ellis
INTRODUCTION
Breast cancers are thought usually to develop from regions of cellular atypia into clinically evident preinvasive or invasive lesions with subsequent evolution of lethal features such as metastatic spead and therapeutic resistance. Biological and statistical models that guide our thinking about breast cancer genomic progression events that underlie this process include linear, stepwise models, such as those proposed by Vogelstein and coworkers for colon cancer (Fig. 24-1A) and branched, Darwinian evolutionary models, such as that proposed by Nowell, where there is no fixed order of events or steps (Fig. 24-1B) (1, 2). The recent application of next-generation sequencing technology has uncovered immense genomic complexity but has mostly focused on a single time point— the breast cancer genome at diagnosis. Longitudinal studies of individual cancer genomes over time will ultimately be required to completely characterize the genomics of breast cancer progression and are now possible as massively parallel sequencing platforms become more accessible. In this chapter, we will describe the theoretical basis for breast cancer evolution, from the earliest stages to advanced disease. We will review what is known about the dynamic structural changes in the genome that underlie the disease.
NEXT-GENERATION DNA SEQUENCING
The revolution in DNA sequencing technology, with the development of next-generation sequencing instruments, has provided a wealth of new data on the genomic evolution of cancer, and it is worthwhile beginning our discussion with an overview of this technology as many of the discussions in this chapter rely on data derived from these “massively parallel sequencing” (MPS) approaches. The development and commercialization of next-generation DNA sequencing instruments began around 2006 and has made DNA sequencing thousands of times faster and considerably cheaper. With this technological advance, projects to sequence large numbers of cancer genomes became feasible. Here, we will mostly focus on an MPS process called “sequencing-by-synthesis,” as most data sets have used Illumina-based technology. The steps in this process involve generating a DNA library from the sample of interest (Fig. 24-2A) and attaching individal DNA molecules to a glass slide called a flow cell. The individual sequences are then amplified into “clusters” to increase signal intensity (Fig. 24-2B). A sequencing reaction is then performed whereby a different colored fluorophor is activated when each of the four different nucleotides are added to the DNA polymer (Fig. 24-2C) (3, 4). These sequential light reactions are captured by a sensitive light detector and the sequences entered into programs that align the sequences to the reference human genome. The process is remarkably efficient, and sequencing throughput is advancing rapidly. Currently more than 100 billion base pairs (Gigabases, Gb) can be generated per instrument run. Other methodologies and companies for next-generation DNA sequencing include the SOLiD system developed by Applied Biosystems, and single-molecule DNA sequencers, which are still under development (3).
A key advantage of the MPS approach is that the analysis begins with an individual DNA molecule. This is fundamentally different than the original Sanger DNA sequencing method, developed over 35 years ago, which averages sequences across millions of DNA molecules and, as a
result, has limited ability to distinguish variations in DNA sequences. Because MPS provides data on the frequency of a DNA mutation within a population, tumor clonality or heterogenity can be inferred from the variant allele frequency, and this provides new means for studying the clonal progression of cancer. Rare alleles can be detected down to a frequency of between 0.1% and 1% depending on the depth of the sequencing instrument and the fidelity of the enzymes used to generate the original clusters on the flow cell.
result, has limited ability to distinguish variations in DNA sequences. Because MPS provides data on the frequency of a DNA mutation within a population, tumor clonality or heterogenity can be inferred from the variant allele frequency, and this provides new means for studying the clonal progression of cancer. Rare alleles can be detected down to a frequency of between 0.1% and 1% depending on the depth of the sequencing instrument and the fidelity of the enzymes used to generate the original clusters on the flow cell.
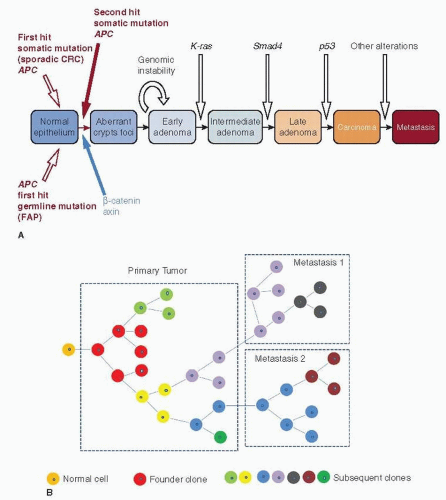 FIGURE 24-1 (A) The development of cancer through sequential steps. This model proposes that colon cancers develop from aberrant foci (atypia) and adenomas in a stepwise process. (From Pinto D, Clevers H. Wnt, stem cells and cancer in the intestine. Biol Cell 2005;97:185-196.) (B) Evolutionary models of cancer with the development of subclones. |
Much of the sequencing funded by the National Cancer Institute has focused on the initial diagnostic sample from invasive cancers through The Cancer Genome Atlas Project, with the goal to catalogue all the somatic mutations and structual abnoramlities in breast cancer (5). The application of this approach in the study of breast cancer is still in its infancy. Our current understanding of the genomic alterations that promote preinvasive to invasive breast cancer is still largely based on comparative genomic hybridization (CGH) techniques, loss of heterozygosity (LOH) analysis, gene expression profiling, and selective gene sequencing studies that have been applied to synchronous preinvasive and invasive breast cancers.
GENOMIC MODELS OF BREAST CANCER PROGRESSION
To understand breast cancer progression, a description of the anatomy of the breast and the histology of preinvasive versus invasive breast cancer is required; the reader is guided to Chapters 1, 9, 21, and 25 which deal with these issues in depth. In brief, several models have been proposed to describe the development of breast cancer that focus on the relationship between preinvasive and invasive breast cancers. The most widely accepted “linear” multistep model suggests a transition from normal epithelium to invasive breast cancer via non-atypical and atypical hyperplasia and in situ carcinoma through accumulation of genetic mutations (6). In this classic Wellings model, premalignant breast lesions arise from terminal ductal lobular units (TDLUs) and give rise to flat epithelial ayplasia (FEA), atypical ductal hyperplasia (ADH), and ductal carcinoma in situ (DCIS), which subsequently progresses, over a long period of time, to invasive ductal carcinoma (IDC), whereas atypical lobular hyperplasia (ALH) and lobular carcinoma in situ (LCIS) progress to invasive lobular cancer (ILC) (Fig. 24-3) (7). This theory was originally based on the histologic observation of the gradual histologic continuity, but it also has been supported by analyses of genetic alterations that compared preinvasive and invasive breast cancers, especially when they occur in the same breast (8, 9, 10 and 11). Subsequent findings introduced the concept of usual ductal hyperplasia (UDH), in which cells pile up to fill the terminal duct (TD) and acini compared to the single or minimally pseudostratified layer of cells in FEA that distends the TDLU, as the direct precursor to ADH (12, 13). However, UDH as the precursor for ADH has not been supported by recent immunohistochemical and molecular evidence. The LOH pattern observed in UDH is notably different from that associated with ADH and DCIS (8, 14, 15, 16, 17 and 18). This linear model of breast cancer progression provided the rationale for detection methods such as mammography in the hope of diagnosing and treating breast cancer at earlier and preinvasive stages before lethal features of the disease have developed (19). However, the occurrence of a preinvasive lesion is probably not an obligatory event in the development of invasive breast cancer. Although many premalignant lesions progress through the lifespan of some patients, others might stay stable throughout their lives; it remains unclear which lesions have the capacity to progress to invasive cancer. The “nonlinear” or “branched” model builds upon the “linear” model in that it agrees that DCIS is the precurser for IDC, but hypothesizes that different grades of DCIS progress to corresponding grades of IDC. In contrast, the “parallel” model hypothesizes that DCIS and IDC are parallel and independent developments from a common progenitor cell through different grades (20). This is supported by the investigation of gene copy number changes in synchronous DCIS and IDC lesions, which demonstrated changes that are specific to DCIS or IDC (21).
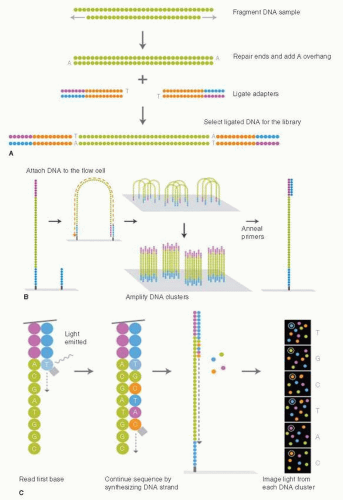 FIGURE 24-2 Next-generation DNA sequencing on the Illumina sequencing instruments. (A) DNA Sample preparation to create the library for sequencing. (B) Attaching the DNA to the surface of the flow cell and generating clusters of identical DNA molecules for sequencing. (C) Sequencing by synthesis. Each round attaches a DNA nucleotide to the DNA molecule and releases light, which is detected by a very sensitive camera. (From Ansourge WJ. Next-generation DNA sequencing techniques. New Biotechnol 2009;25(4):195-203.) |
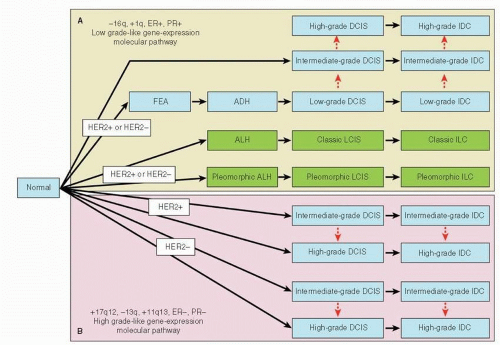 FIGURE 24-3 Multistep human breast cancer progression model. The low grade-like gene-expression molecular pathway is characterized by chromosome 16q loss, predominant ER and PR expression, and a low-grade gene expression profile populated with genes associated with ER positivity. (A) This low-grade pathway is observed in preinvasive lesions of both ductal and lobular subtype. (B) The high grade-like gene-expression molecular pathway is characterized by loss of chromosome 13q; gain of 11q13 and/or amplification of 17q12; infrequent expression of ER and PR; and a high-grade gene expression signature populated with genes associated with cell cycle, centrosomal function, and DNA repair. Pleomorphic atypical ductal hyperplasia (ALH), pleomorphic lobular carcinoma in situ (LCIS), and pleomorphic invasive lobular carcinoma (ILC) phenotypically resemble highgrade tumors, and immunohistochemical (ER positivity) and genetic data (16q loss and 1q gain) support an evolutionary association with the low grade-like gene expression molecular pathway. (From Sgroi DC. Preinvasive breast cancer. Annu Rev Pathol 2010;5:193-221.) |
GENOMIC ALTERATIONS SUPPORTING PREINVASIVE LESIONS AS PRECURSORS OF INVASIVE BREAST CANCER
The molecular differences among the preinvasive and invasive breast cancers are largely unknown but have been an area of great research interest with the hope to identify the key events that drive the development and progression of invasive breast cancer. The pathological heterogeneity and the microscopic size of the preinvasive lesions have posed a practical challenge in isolating sufficient material that is devoid of contaminating tissues. The availability of laser capture microdissection (LCM) and genome-wide analysis tools provide a new opportunity to discover genetic events specifically activated or inactivated in the course of breast cancer development.
LOH and CGH Studies
Multiple studies indicate that genetic alterations that confer the potential for invasive growth already exist in the earliest phenotypically recognized preinvasive stages. Initial studies of the genetic evolution of breast cancer progression used relatively insensitive loss of heterozygosity (LOH)/comparative genomic hybridization (CGH) techniques (8, 10, 22, 23). For example, O’Connell et al. studied 399 microdissected preinvasive lesions (211 UDH, 51 ADH, 81 noncomedo DCIS, and 56 comedo DCIS) for LOH at 15 polymorphic genetic loci known to exhibit high rates of loss in invasive breast cancer (IBC) and assessed the sharing of LOH between synchronous preinvasive and invasive cancers. For breast samples without DCIS and IBC, 37% of UDH and 42% of ADH lesions showed LOH in at least one locus, although loss at any given locus was uncommon (range, 0%-15%), suggesting that the development of hyperplasias can involve many different genes. In breast samples without IBC, LOH was common in DCIS, with 70% of noncomedo lesions and 79% of comedo lesions showing at least one loss with up to 37% of samples harboring LOH on chromosomes 16q, 17p, and 17q. When DCIS lesions from breasts with or without IBC were compared, substantially more LOH was observed in the breast with IBC at a few loci (on chromosomes 2p, 11p, and 17q), suggesting that genetic alterations in these regions may be important in the progression of DCIS to invasive disease. Among specimens harvested from breasts with IBC, 37% of concurrent UDH, 45% of ADH, 77% of noncomedo DCIS, and 80% of comedo DCIS lesions shared LOH with synchronous cancers at one locus or more. Similarly, in another CGH study performed on a panel of breast tumors that included 10 DCIS, 18 IBC, and two lymph node metastasis, there was an overall trend toward an increase in the number of genetic gains and losses in the IBC (24). In a study of 41 cases of sporadic breast cancer that focused on LOH of chromosome 11q13, LOH on chromosome 11q13 was present in 24 of 36 (67%) of the informative invasive breast cancer cases. The identical allelic loss was shared in the microdissected DCIS and the corresponding invasive breast cancer in 71% (15 of 21) of the available cases (23). Moelans et al. analyzed 21 genes including transcription factors and thyrosine kinases in DCIS and adjacent IDC (25) and found that there were no copy number differences between them. These studies provided molecular genetic support for the notion that invasive breast cancer arises from preinvasive lesions and that DCIS is genetically as advanced as IDC and the driving genetic events already occurred at the preinvasive stage. This conclusion is thus somewhat paradoxical because DCIS is a benign disorder and invasive disease is not. We therefore still do not have a clear idea of the genomic determinants of the DCIS to invasive transition, which is a clear impetus for more detailed studies using MPS.
Expression Profiling
The molecular similarity between preinvasive lesions and invasive breast cancer has also been observed at the level of gene expression. Using LCM, T-7 based RNA amplification and DNA microarrays analysis, Ma et al. compared the gene expression profile of normal TDLU epithelium and synchronous ADH, DCIS, or IDC in a study of 36 breast cancer specimens (26). Compared to patient-matched normal epithelial cells, significant alterations in global gene expression occurred at ADH, which persisted in the later stages of DCIS and IDC. There were extensive similarities at the transcriptome level among the paired ADH, DCIS, and IDC without any consistent gene expression unique to each of the three identities. These observations were consistent with an earlier study of global gene expression profiles using serial analysis of gene expression (SAGE), although performed on a limited cohort of normal mammary epithelial cells, DCIS, IDC, and metastatic disease (27).
Similar to IBC, DCIS exhibits significant histologic and biological diversity between different cases. Under microarray gene expression analysis, intrinsic subtypes originally described for IBC have also been observed in DCIS (28, 29 and 30). In a recent immunohistochemistry (IHC) analysis of a tissue microarray composed with 188 cases of pure DCIS (31), a frequency of 38.3% for Luminal A (ER+/PR+/HER2-), 6.9% Luminal B (ER+ and PR- and/or HER 2+), 14.9% HER2 (ER-/PR-/HER2+), 7.5% TN (ER-/PR-/HER2-) and 4.2% basallike (ER-/PR-/HER2-/CK5/6 and/or EGFR+) was observed according to IHC criteria of intrinsic subtypes (32). These studies indicated that the molecular heterogeneity of IBC is reflected at the stage of DCIS, and DCIS may be classified in a manner similar to invasive breast cancer.
GENETIC ALTERATIONS INDICATING DISTINCT GENOMIC PATHWAYS ASSOCIATED WITH LOW- AND HIGH-GRADE BREAST CANCERS
In gene expression profiling studies of synchronous DCIS and IDC lesions, the greatest alterations were among the different histological grades of DCIS and IDC (26). Notably, the grade I and grade III tumors demonstrated reciprocal gene expression patterns, whereas grade II tumors exhibited a hybrid pattern of grade I and grade III signatures. ADH samples demonstrated a grade I gene expression signature and clustered with the low-grade DCIS and IDC. Similarly, several comparative genomic hybridization studies revealed that the low-grade and high-grade DCIS have distinct gains and losses of genetic material. In the CGH study by Buerger et al. on 38 DCIS and 6 associated invasive breast cancers,
losses of 16q were exclusively seen in well- and intermediately-differentiated DCIS. A higher frequency of gains of 1q and losses of 11q was observed in intermediately-differentiated DCIS. The poorly-differentiated DCIS displayed complex genomic alterations, including loss of 8p, 11q, 13q, and 14q and gains of 1p, 8q, and 17q. This was characterized by a higher frequency of amplifications of 17q12 and 11q13. Analysis of paired IBC revealed a CGH pattern similar to the DCIS counterpart (33). This data provided further evidence that DCIS are precursor lesions of IBC, but progression from low- and high-grade DCIS to IBC are likely from different genetic pathways (34) (Table 24-1).
losses of 16q were exclusively seen in well- and intermediately-differentiated DCIS. A higher frequency of gains of 1q and losses of 11q was observed in intermediately-differentiated DCIS. The poorly-differentiated DCIS displayed complex genomic alterations, including loss of 8p, 11q, 13q, and 14q and gains of 1p, 8q, and 17q. This was characterized by a higher frequency of amplifications of 17q12 and 11q13. Analysis of paired IBC revealed a CGH pattern similar to the DCIS counterpart (33). This data provided further evidence that DCIS are precursor lesions of IBC, but progression from low- and high-grade DCIS to IBC are likely from different genetic pathways (34) (Table 24-1).
TABLE 24-1 Chromosomal Aberrations of DCIS and IDC | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||
In addition, FEA and ADH has morphological overlap with low-grade carcinomas, and ADH is accepted as a precursor lesion for low-grade/noncomedo DCIS. Molecularly, FEA has a genetic profile that overlaps with those of synchronous lowgrade DCIS and low-grade invasive carcinomas and has a high rate of LOH at 16q, supporting its precursor role in the evolution to low-grade cancers (11). In another study, FEA exhibited recurrent chromosomal copy number gains and losses (gains on 15p, 16p, and 19; losses on 16q, 17p, and X), which overlap with those observed in both ADH and low-grade DCIS (9).
However, the precursor lesion of high-grade/comedo DCIS is not clear. High-grade DCIS lesions are associated with more complex genetic alterations and overexpression of genes related to mitotic activity and cell cycle processes (35). In a comparison of copy number changes of 21 breast cancer-related genes between laser-microdissected DCIS and adjacent IDC lesions (25), no significant differences existed between DCIS and adjacent IDC. However, low/intermediate-grade DCIS showed on average 6 gains/amplifications versus 8 in high-grade DCIS. Furthermore, alterations of AURKA (aurora kinase A) and CCNE1 (cyclin E1) were exclusively found in high-grade DCIS, and HER2, PRDM14 (PR domain containing 14), and EMSY amplification was more frequent in high-grade DCIS than in low/intermediate-grade DCIS. These data indicate that DCIS is genetically as advanced as IBC and support a model in which different histological grades of DCIS are associated with distinct genomic changes that progress to IDC in different routes.
EVOLUTION FROM LOWER TO HIGHER GRADE LESIONS
The significant intratumoral histological and biological diversity within cases of DCIS argues for an evolution of DCIS from lower grade to higher grade (28). In an analysis of 120 consecutive cases of pure DCIS, 45.8% of cases showed areas of diversity in nuclear grades, including 30% with grades I and II, 6.6% with grades II and III, and 9.2% with grades I, II, and III. In addition, about one-third of the cases showing histologic diversity also showed biologic diversity for one or more biomarkers that included ER, GATA3, HER2, CK5/6, CK18, and p53 by immunohistochemistry staining. Similarly, in studies assessing the LOH, DCIS contained many of the same specific genetic defects regardless of histologic differentiation, although the absolute number of defects was found to be higher in higher grade lesions (8). Furthermore, a significant subset of genes expressed at higher levels in grade III DCIS compared to grade I DCIS were further elevated in IDC. In addition, the link between tumor grade and transition from DCIS to IDC is consistent with the clinical observation that grade III DCIS is more likely to be associated with occult invasive disease than grade I DCIS (36).
MOLECULAR MARKERS OF DCIS THAT PREDICT RECURRENCE AND INVASIVE PROGRESSION
The evolution of genetic events that drives the process of breast tumorigenesis is poorly understood. As demonstrated in previous studies, many genetic alterations present in the invasive breast cancer already exist in the earliest phenotypically identifiable lesions such as ADH. However, gene expression profiling, CGH, and candidate gene approach, which are the main technologies used in these studies, are limited in their capacity for detailed genomic interrogation. A comprehensive genomic sequencing study of synchronous preinvasive and invasive cancer in comparison to the normal breast epithelial cells is needed but has not been reported. In this section, we will focus on available data investigating molecular markers that may predict the prognosis of DCIS and potential candidate drivers in cancer progression.
Prognostic Markers
Low ER or PR expression or HER2 amplification is associated with higher grades of DCIS and recurrence (37, 38, 39, 40 and 41). A molecular signature of lack of ER and PR, HER2 overexpression, accumulation of p53, and high Ki67 expression was proposed to predict recurrence (42). COX-2 and p16 have also been associated with progression or recurrence. In a retrospective study, DCIS lesions that were positive for p16, COX-2, and Ki67 expression were significantly associated with risk of subsequent invasive cancer, whereas DCIS lesions that either lacked ER but were positive for HER2 and Ki67 or that lacked COX2 but were positive for p16 and Ki67 were associated with recurrence of DCIS (43). However, with the exception of ER/PR, none of these molecular markers are routinely assessed in the clinic due to the lack of sufficient evidence or established interventions.
Several other molecular markers, including cell cycle regulation and apoptotic markers (cyclin D1, cyclin A, cyclin E, p21, p27, p53, Bcl-2, Bax, Survivin, c-myc, and Rb), angiogenesis-related proteins (VEGF and heparanase-1), and extracellular matrix-related proteins (CD10, secreted protein acidic and rich in cysteine), have been investigated in molecular epidemiology studies; however, the data have not been conclusive (44).
Candidate Drivers of Invasive Progression
ER is commonly expressed in preinvasive lesions, 95% and 75% in ADH/LCIS and DCIS lesions, respectively (10). Binding
of estrogen to ER stimulates the growth and differentiation of breast epithelium; therefore, prolonged estrogen exposure in preinvasive disease might have a role in the development of breast cancer. Consistent with the important role of estrogen in breast cancer progression, tamoxifen has been shown to be an effective drug for prevention of breast cancer in high risk patients as well as an effective adjuvant hormonal therapy for patients with resected ER+ DCIS.
of estrogen to ER stimulates the growth and differentiation of breast epithelium; therefore, prolonged estrogen exposure in preinvasive disease might have a role in the development of breast cancer. Consistent with the important role of estrogen in breast cancer progression, tamoxifen has been shown to be an effective drug for prevention of breast cancer in high risk patients as well as an effective adjuvant hormonal therapy for patients with resected ER+ DCIS.
HER2 overexpression occurs commonly in high-grade lesions (60%) compared to low-grade lesions (10%). In addition, HER2 is not overexpressed in TDLUs, very rarely in ADH, and about 2% in LCIS. The absence of HER2 overexpression in the earliest phase of preinvasive disease and its association with higher grade DCIS and more aggressive clinic behavior suggest HER2 overexpression is a driving event in cancer progression (45). A randomized phase III trial of adjuvant radiotherapy with or without trastuzumab in patients with HER2+ DCIS resected by lumpectomy is ongoing to evaluate the effect of HER2-targeting in ipsilateral breast cancer recurrence (NCT00769379).
As mentioned above, the precise genetic event(s) triggered during the transition from DCIS to IDC is a critical unknown in the study of breast cancer. A few limited studies that compared the IDC with the adjacent DCIS suggested that c-Myc or FGFR1 amplification may be involved in this process because these genetic events occurred more frequently in the IDC compared to the adjacent DCIS lesions (46, 47 and 48), while PIK3CA, AKT1, and TP53 mutations are early events that appeared to already exist at the DCIS stage (Table 24-2). Knudson et al. confirmed in their study that DCIS present in concert with IBC harbors gene expression profiles similar to IBC; however, when IBC and pure DCIS were compared, the expression differences became clearer (49). Genes associated with epithelial-to-mesenchymal transition and myoepithelial specific genes were enriched in IBC relative to DCIS, particularly in the stromal component. There have been few in vivo studies addressing the function of genes in the progression of DCIS to IDC. Using a “mammary intraductal DCIS” xenograft model, Lee et al. studied the progression of DCIS to invasive breast cancer in vivo by introducing specific genes in the human DCIS cell line. Four genes, including a protease inhibitor (CSTA) and three genes involved in cell adhesion and signaling (FAT1, DST, and TMEM45A), which were usually elevated in clinical samples of DCIS, were found to suppress the progression of DCIS to invasive cancer (50).
Related posts:
 Breast Cancer Screening
Breast Cancer Screening
 Ductal Carcinoma In Situ and Other Intraductal Lesions: Pathology, Immunohistochemistry, and Molecular Alterations
Ductal Carcinoma In Situ and Other Intraductal Lesions: Pathology, Immunohistochemistry, and Molecular Alterations
 Adjuvant Systemic Therapy: Endocrine Therapy
Adjuvant Systemic Therapy: Endocrine Therapy
 Preoperative Endocrine Therapy for Operable Breast Cancer
Management Summary for the Care of Patients with Metastatic Breast Cancer
Nursing Care in Patient Management and Quality of Life
Preoperative Endocrine Therapy for Operable Breast Cancer
Management Summary for the Care of Patients with Metastatic Breast Cancer
Nursing Care in Patient Management and Quality of Life
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


