Abstract
The rare autoimmune polyglandular failure syndromes (APS) comprise a juvenile (APS1) and an adult type (APS2 and 3). APS1 is caused by mutations in the autoimmune regulatory ( AIRE ) gene on chromosome 21 and is inherited in an autosomal recessive manner. Mutations in the AIRE gene impair self-tolerance and result in defect proteins causing autoimmune destruction of target organs. Genetic testing identifies patients with APS1. For APS2/3 disease susceptibility genes are the human leucocyte antigen on chromosome 6, the cytotoxic T lymphocyte antigen (chromosome 2), the protein tyrosine phosphatase nonreceptor type 22 (chromosome 1), the forkhead box P3 (X chromosome), and the interleukin-2 receptor alpha gene region (chromosome 10). These genes are involved in the immune regulation and T-cell activation foremost within the immunological synapse. Further candidate genes with joint risk for autoimmune thyroid disease and type 1 diabetes (APS3) are the v-erb-b2 erythroblast leukemia viral oncogene homolog 3 gene on chromosome 12 and the C-type lectin domain family 16 member A on chromosome 16. The latter one might be involved in pathogen recognition.
Keywords
genetics, autoimmune polyglandular failure syndrome (APS), APECED, AIRE1
Definition, incidence, prevalence
The autoimmune polyglandular failure syndromes (APS), also known as multiple endocrine abnormalities (MEA), define the autoimmune-induced failure of at least two glands, and comprise a wide spectrum of autoimmune disorders. They encompass a rare juvenile type (APS1) and more frequent adult types (APS2 and APS3). APS1 is also known as autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) because it is formed by three main disorders, which are chronic mucocutaneous candidiasis, autoimmune hypoparathyroidism, and autoimmune Addison’s disease, AD. In contrast to APS1, APS2 and APS3 primarily manifest in adult age. APS2 is defined as the association of autoimmune AD and a further autoimmune endocrine disorder. The APS3 variant is defined by the presence of autoimmune thyroid disease, AITD, and type 1 diabetes, T1D. In contrast to APS1, chronic candidiasis is not present in APS2 and APS3. Further autoimmune endocrine and nonendocrine component disorders may occur in all APS types. Due to the tremendous overlap of phenotypes in APS2 and APS3, for daily use, it is clinically relevant to differentiate the more common adult type encompassing both APS2 and APS3 from the rare juvenile type APS1.
APS1 manifests in infancy or early childhood. The female-to-male ratio approximates one. Prevalence of APS1 is generally rare: 2–3:1,000,000 in Great Britain, but it occurs more frequently in three genetically isolated populations who are Finnish 1:25,000, Iranian Jews 1:9000, and Sardinians 1:14,000. Prevalence in other populations is 1:43,000 in Slovenia, 1:80,000 in Norway, and 1:129,000 in Poland. The adult type APS2/3 is more common, but still a rare syndrome. Its prevalence is 1:20,000. It occurs more frequently in women. The male-to-female ratio is 1:3. The incidence of APS2/3 peaks at ages 20–60 years, mostly in the third or fourth decade.
Clinical spectrum
APS1 is characterized by three major disease components. These are chronic mucocutaneous candidiasis (chronic susceptibility to candida yeast infection), autoimmune hypoparathyroidism (parathyroid gland failure, which affects calcium metabolism including nails and tooth enamel), and AD (autoimmune adrenal failure). The first manifestation occurs in infancy or early childhood and is typically mucocutaneous candidiasis. The second manifestation is hypoparathyroidism around age seven years and the third manifestation is AD around age 13 years. Clinical presentation of the diseases varies and comprises further endocrine and nonendocrine minor component disorders. These include autoimmune endocrinopathies (foremost primary hypogonadism, T1D and AITD are less prevalent), gastrointestinal disorders with malabsorption, pernicious anemia, autoimmune hepatitis, autoimmune skin disorders (vitiligo, alopecia), ectodermal dysplasia (dental enamel hypoplasia, nail dystrophy), and keratoconjunctivitis. The minor components of the disorder do not manifest until the fifth decade of life. Clinical diagnosis of APS1 requires the presence of two of the three major component diseases: AD and/or primary hypoparathyroidism, and/or chronic mucocutaneous candidiasis. Most patients have four disease manifestations and may display up to 10 component diseases. Some patients have the full constellation of diseases, while others do not. However, in some patients, primary hypoparathyroidism was the only manifestation.
APS2 is characterized by the presence of autoimmune adrenalitis (AD) and at least one other autoimmune endocrine disorder. The second disease component may either be primary hypogonadism or AITD (Graves’ disease, GD, or Hashimoto’s thyroiditis, HT) or T1D, or both. Further endocrine (primary hypogonadism, primary hypoparathyroidism) and nonendocrine component diseases (pernicious anemia, autoimmune hepatitis, alopecia, vitiligo, celiac disease with malabsorption, and myasthenia gravis) may be present. The associated minor autoimmune diseases are less frequent than in APS1. APS2 mostly occurs in adulthood during the third and fourth decades. About half of patients with APS2 initially have T1D developing additional AD (APS2) within a few years. In adults, the manifestation of one autoimmune endocrine disorder increases the risk of developing other autoimmune disorders while several years may separate the onset of different component diseases. Diseases resulting in autoimmune-induced tissue destruction have a prolonged phase of cellular loss preceding overt autoimmune glandular failure. Silent autoantibodies are prevalent in families with APS2 and antibody screening is predictive for the early diagnosis of adrenal failure, especially in children.
The APS3 variant is defined by the presence of AITD and T1D without adrenal involvement. Nonendocrine component diseases include type A autoimmune gastritis, pernicious anemia, vitiligo, alopecia, myasthenia gravis, Sjögren’s syndrome, systemic lupus erythematosus, and rheumatoid arthritis. AITD peaks in the fourth decade for GD or fifth and sixth decade for HT. The simultaneous occurrence of autoimmune induced hypothyroidism (HT) and T1D is often accompanied by hypoglycemia due to decreased insulin requirement and increased insulin sensitivity. Glucose intolerance accompanies autoimmune hyperthyroidism in 50% of patients. In contrast to patients with APS1, patients with adult APS do not develop mucocutaneous candidiasis. Also, primary hypoparathyroidism, which is indicative of APS1, is rare in APS2/3. In adult APS, circulating organ-specific autoantibodies are present in each of the component diseases. Occasionally, antibodies will cross-react with more than one gland (e.g., steroid-producing cells). Antithyroid peroxidase and antiparietal cell antibodies are prevalent in healthy relatives of APS patients. Antibodies usually precede clinical disease; however, in contrast to anti-islet antibodies, antithyroid antibodies can be present for decades without progression to overt disease. Antibodies against steroidal enzymes, for example, 21-hydroxylase, are of high predictive and prognostic value. They will aid identifying patients at risk for developing AD.
Patients with the adult APS could be exposed to many limitations of their illness in daily life. To objectify the degree of physical and emotional distress, the psychometric profile of patients with APS2/3 was prospectively evaluated and happened to be severely impaired. Treatment modalities that would improve their well-being are warranted. In summary, current diagnosis of adult APS involves serological measurement of organ-specific autoantibodies and subsequent functional testing. Management of patients with APS, including their family relatives, is best performed in centers with special expertise in autoimmune endocrine disorders.
Genetic pathophysiology
Known Mutations/Polymorphisms and Specific Phenotypes
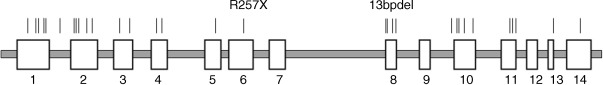
APS1 is a monogenic disease due to a defect in a single gene. Haplotype analyses suggest that APS1 is caused in different populations by a number of different mutations in a single gene. This gene is the autoimmune regulatory ( AIRE ) gene on chromosome 21, location 21q22.3. The gene was identified in 1997 by positional cloning. The AIRE gene is approximately 13 kb in length and the coding sequence comprises 14 exons. Currently, more than 50 different mutations in the AIRE gene causing APS1 have been detected. These mutations are distributed over the whole coding region of the gene ( Fig. 25.1 ). They comprise point mutations (nonsense and missense mutations), insertions, and deletions resulting in frameshifts and splice site mutations. The most frequent mutations are R257X, exon 6 and 967-979del13bp (also denominated in the literature as 1094-1106del13bp, exon 8). The R257X mutation is characterized by a C→T substitution at position 769 of the AIRE gene, resulting in a TGA codon (stop codon) instead of CGA (coding for arginine), leading to a truncated regulator protein. It is present in 83% of Finnish APS1 patients and predominates in Italian and Polish APS1 patients. Also, it is found in APS1 patients from Great Britain, Germany, France, Sweden, the Netherlands, Switzerland, Austria, Hungary, Croatia, Serbia, Slovenia, Russia, the USA (Caucasians), and New Zealand. It was detected in patients on different chromosomal haplotypes suggesting various mutational origins. The frequent 13-bp deletion accounted for 70% of British and 53% of North American (Caucasian) APS1 alleles but also occurred in Finland, Sweden, Norway, the Netherlands, Germany, Italy, Hungary, Canada, New Zealand, Russia, and other countries. Thus, both R257X and 967-979del13bp have been noted in patients of different geo-ethnic origins, and both were associated with multiple different haplotypes using closely flanking polymorphic markers showing likely multiple mutation events. The mutations causing APS1 are inherited in an autosomal recessive way. One mutation (G228W) in the AIRE gene observed in an Italian family has a dominant inheritance. In this family, only one heterozygous mutation was found in the entire coding sequence of the AIRE gene.
In contrast to APS1, APS2 and 3 are genetically complex and multifactorial syndromes. Several genetic loci possibly interact with environmental factors. Adult APS types are strongly associated with certain alleles of the human leucocyte antigen ( HLA ) genes within the major histocompatibility complex (MHC) ( Table 25.1 ). The HLA-DRB1*03 allele was strongly increased in patients with adult APS (50.7%) versus both controls (21.8%, P < 0.0001; RR 2.32, 95% CI 1.62–3.33) and monoglandular autoimmune disease (11.4%, P < 0.0001). HLA-DRB1*03 was highly prevalent in APS2/3 patients with early versus late disease onset ( P < 0.05, logistic regression analysis). HLA-DRB1*04 allele carriers were more present in adult APS versus controls (53.4% vs. 22.4%, P < 0.0001; RR 2.38, 95% CI 1.68–3.38). Further, HLA-DQB1*02 was increased in adult APS versus controls ( P < 0.01), whereas HLA-DQB1*06 was decreased ( P < 0.001). Thus, HLA-DRB1*03 is a stronger genetic marker in adult APS than in monoglandular autoimmune diseases, foremost in those with early disease onset.
| Gene locus | Chromosomal location | Exons | Mutations/polymorphisms |
|---|---|---|---|
| IMMUNE-MODIFYING GENES | |||
| MHC | 6p21 | − | MHC class II region |
| MICA | 6p21.3 | 6 exons | Exon 5: GCT repeat (transmembrane region) |
| CTLA-4 | 2q33 | 4 exons | Exon 1: SNP +49 A/G STR (AT) n repeat within 3′UTR, allele 106 Intron 1: SNP +1822 C/T 3′UTR: SNPs CT60/G, JO31/G, JO30/G |
| CD40 | 20q11 | 9 exons | SNP C/T (position-1, designated as CD40-E1SNP) |
| CYTOKINE-RELATED GENES | |||
| IL-1RA | 2q14.2 | 6 exons | Intron 2: VNTR * |
| IL-4 | 5q31.1 | 4 exons | Promoter-590 C/T |
| TNF-α | 6p21.3 | 4 exons | Promoter-863 C/A, 1031 T/C |
| Autoimmune regulator gene AIRE-1 | 21q22.3 | 14 exons | >45 mutations, most frequent mutations: Exon 6: R257X, Arg257stop Exon 8: 13bpdel, deletion of 13 bp (964del13) |
* A penta-allelic 86-bp tandem repeat (VNTR) occurs in intron 2, of which allele 2 (IL1RN*2) is associated with autoimmune conditions.
APS2 frequently clusters in families. Several generations are often affected by one or more component diseases. The inheritance pattern is autosomal dominant with incomplete penetrance in some patients. Two genes have been shown to be associated with APS2. These are HLA genes on chromosome 6 and the cytotoxic T lymphocyte antigen ( CTLA-4 ) gene on chromosome 2 (location 2q33). Of these, HLA appears to have the strongest gene effect. Patients with APS2 had significantly more the HLA alleles DRB1*03 ( P < 0.0001), DRB1*04 ( P < 0.000005), DQA1*03 ( P < 0.0001), DQB1*02 ( P < 0.05) when compared to controls. Less frequent in APS2 were DRB1*15 ( P < 0.05), DQA1*01 ( P < 0.0005), DQB1*05 ( P < 0.005). With regard to frequency and linkage of these alleles, the susceptible haplotypes DRB1*0301-DQA1*0501-DQB1*0201 and DRB1*0401/04-DQA1*0301-DQB1*0302 were deduced. Protective haplotypes in this study were DRB1*1501-DQA1*0102-DQB1*0602 and DRB1*0101-DQA1*0101-DQB1*0501. Patients with T1D as a singular disease had the same susceptible and protective HLA alleles and haplotypes. The prevalence of DRB1*03 and DRB1*04 in APS2 patients was not due to the presence of diabetes, since the APS2 without T1D had the same allele distribution. These data suggest a common immunogenetic pathomechanism for T1D and APS2, which might be different from the immunogenetic pathomechanism of other autoimmune endocrine diseases.
Many APS2 component disorders are associated with an increased frequency of the HLA haplotype A1, B8, DR3, DQA1*0501, DQB1*0201. AD is strongly associated with DR3 and DR4 ; the observed relative risks are 6.0, 4.6, and 26.5 for DR3, DR4, and DR3/DR4, respectively. AD is also correlated with DQ2/DQ8 with DRB1*0404, both as a single disease as well as within APS2. T1D is positively associated with DR4-DQB1*0302, DRB1*04-DQA1*0301-DQB1*0302, or DRB1*03-DQA1*0501-DQB1*0201 (DR3-DQ2), and negatively associated with DRB1*15-DQA1*0102-DQB1*0602. In APS2 patients without islet cell autoimmunity, only the haplotype DR3-DQB1*0201 occurred more frequently. The DRB1*04-DQB1*0301 haplotype increases the risk for developing HT.
Very recently, the HLA class II alleles, haplotypes, and genotypes were determined in a large cohort of patients with adult APS, AITD, T1D, and in healthy controls by the consistent application of high resolution typing at a four digit level. Inclusion of family members of APS patients enabled the assignment of segregation-derived HLA class II haplotypes. Comparison of the allele and haplotype frequencies significantly discriminated patients with APS versus AITD and controls. The alleles DRB1*03:01, *04:01, DQA1*03:01, *05:01, and DQB1*02:01, *03:02 were more prevalent ( P < 0.001) in adult APS than in AITD and controls. The DRB1*03:01-DQA1*05:01-DQB1*02:01 (DR3-DQ2) and DRB1*04:01-DQA1*03:01:DQB1*03:02 (DRB1*04:01-DQ8) haplotypes were overrepresented in adult APS ( P < 0.001). The combination of both haplotypes to a genotype was highly prevalent in adult APS versus AITD and controls ( P < 0.001). Dividing the APS collective into those with AD and T1D (APS2) and those without AD but including T1D and AITD (APS3) demonstrated DR3-DQ2/DRB1*04:01-DQ8 as a susceptibility genotype in APS3 ( P < 0.001), whereas the DR3-DQ2/DRB1*04:04-DQ8 genotype correlated with APS2 ( P < 0.001). The haplotypes DRB1*11:01-DQA1*05:05-DQB1*03:01 and DRB1*15:01-DQA1*01:02-DQB1*06:02 were protective in APS3 but not in type 2 ( P < 0.01). Thus, the association of HLA class II haplotypes with APS2/3 depends on the contribution of either T1D or AD. Susceptible haplotypes favor the development of polyglandular autoimmunity in AITD patients.
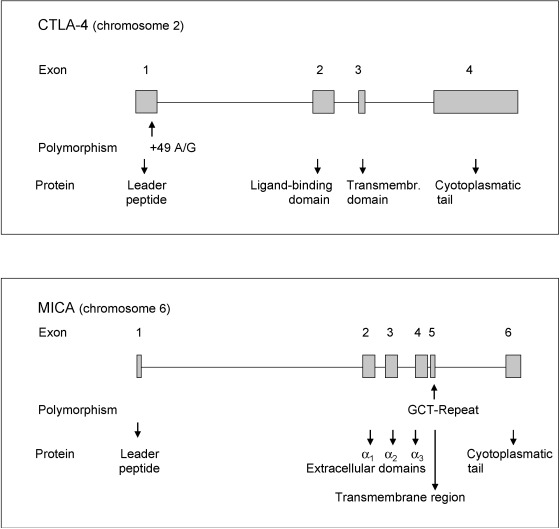
The CTLA-4 gene is assigned to chromosome 2 (location 2q.33). It comprises four exons and encodes a protein that acts as an important modifier of T-cell activation with downregulatory properties. An A49G substitution in exon 1 of the CTLA-4 gene with more G alleles has been associated with GD in Caucasians and Asians and with HT. With respect to GD, family studies give evidence for an increased transmission of the G allele from heterozygous parents to affected offspring compared to unaffected offspring. A 3′ microsatellite (AT) n repeat of the CTLA-4 gene is relevant. The 106 bp allele of the AT repeat was more frequently observed in Caucasian patients with GD than in healthy controls. CTLA-4 alleles have been linked to AITD, and to a weaker extent to T1D. AD was also associated with CTLA-4 alleles, particularly in a subgroup showing HLA -DQA1*0501. Data indicate interactions between HLA and CTLA-4 genes, further unidentified genes and environmental factors.
APS3 is also characterized by a complex inheritance pattern. Family and population studies showed that the APS3 variant syndrome has a strong genetic background. Whole genome and candidate gene approaches identified several gene variations that are present in both AITD and T1D. Most important common disease susceptibility genes are HLA , CTLA-4 , the protein tyrosine phosphatase non-receptor type 22 ( PTPN22 on chromosome 1), the forkhead box P3 ( FOXP3 on the X chromosome) and the interleukin-2 receptor alpha (IL-2Ralpha)/CD25 gene region (chromosome 10), all of them contributing to the susceptibility for APS3. With respect to the underlying pathogenetic mechanisms, these genes are altogether involved in the immune regulation, in particular in the immunological synapse and T-cell activation. In addition to these common genes, there are further candidate genes with joint risk for AITD and T1D, in particular the v-erb-b2 erythroblast leukemia viral oncogene homolog 3 ( ERBB3 ) gene on chromosome 12 and the C-type lectin domain family 16 member A ( CLEC16A on chromosome 16). The latter one might be involved in pathogen recognition.
HLA class II is a potential gene locus for combined susceptibility to T1D and AITD as has been shown in Caucasians and Asians. Most family studies gave evidence that the haplotype HLA -DR3-DQB1*0201 is the primary haplotype conferring susceptibility to both T1D and AITD within families. Here, DR3 seems to be the primary allele conferring risk to both T1D and AITD, whereas DQB1*0201 is less relevant. Many population studies indicate that both HLA haplotypes DR3-DQB1*0201 and DR4-DQB1*0302 contribute to the APS3 variant of combined T1D and AITD.
The causative CTLA-4 gene polymorphism for autoimmunity may be located in the 3′UTR (untranslated region) of the CTLA-4 gene. Here, an (AT) n microsatellite polymorphism occurs with longer and shorter repeats of AT ( Fig. 25.2 ). The longer repeats are associated with decreased inhibitory function of CTLA-4 . Longer repeats were correlated with a shorter half-life of the CTLA-4 mRNA than shorter repeats. CTLA-4 CT60, another CTLA-4 gene polymorphism, was analyzed in patients with APS3, AITD, T1D, and healthy controls. The CT60 G/G genotype was significantly more common in patients with APS3 than in healthy controls (48.6% vs. 32.0%, OR 2.01, 95% CI 1.07–3.77, P = 0.038). The CT60 allele frequencies differed as well between APS3 patients and controls, with the predisposing G allele being increased in APS3 (OR 1.63, 95% CI 1.03–2.55, P = 0.042). Patients with APS3 did not differ from those with AITD ( P = 0.602) or T1D ( P = 0.362).
The PTPN22 gene maps on chromosome 1, location 1p13. This gene encodes the lymphoid tyrosine phosphatase (LYP) protein. Alternative splicing of this gene results in two transcript variants encoding distinct isoforms of the protein. A single nucleotide polymorphism (SNP) in the PTPN22 gene, an 1858C→T transition, results in an arg620-to-trp (R620W) substitution in the LYP protein. The minor T allele was found to be associated with T1D, AITD, and other autoimmune diseases. This is involved in altered T lymphocyte activation. In Asian patients, a novel SNP in the promoter region of the PTPN22 gene, G1123C, has been recently identified and associated with T1D and AITD. Therefore, the promoter SNP is a further possible causative variant for endocrine autoimmunity. Additional candidate polymorphisms may be also causative.
In an association study, 310 white subjects with APS3, AITD, T1D, or healthy controls were genotyped for the C1858T polymorphism. All subjects were also typed for HLA-DRB1. The PTPN22 1858 minor T-allele frequency was strongly increased in patients with APS3 (23.6%) compared with controls (8.0%, P < 0.001), with patients with AITD only (8.6%, P < 0.006), or with T1D only (10.7%, P < 0.028). T-allele carriers were also more frequently present in the group with APS3 vs. controls (41.4% vs. 14.0%, OR 4.35, 95% CI 2.08–9.09), AITD (17.1%, OR 3.42, 95% CI 1.56–7.48), and T1D (21.4%, OR 2.59, 95% CI 1.23–5.45). Especially in subjects with HT + T1D, T-allele carriers were mostly frequent (50% vs. 14%, OR 6.14, 95% CI 2.62–14.38, P < 0.001). Considering all included patients with AITD, T-allele carriers were 29.3% vs. 14.0% in controls ( P < 0.008, OR 2.54, 95% CI 1.30–4.98). Patients carrying the PTPN22 1858 T allele had a twofold increased frequency of the HLA-DRB1*03 allele (64.7% vs. 37.3%, P < 0.034). In conclusion, the PTPN22 gene is a joint susceptibility locus for joint AITD (especially HT) and T1D (APS3).
Data regarding polymorphisms of immunoregulatory genes in polyglandular failure are lacking. Recently, the putative association between a polymorphism of the proinflammatory cytokine gene tumor necrosis factor TNF-α -308 and mutations of the AIRE gene with APS in adults was analyzed. The TNF-α -308*A allele occurred more frequently in patients (0.269) than in controls (0.163, P = 0.008). Also, TNF-α -308*A carriers were more frequent in patients than controls (47.8% vs. 31.1%, OR 1.89, 95% CI 1.19−3.00). The frequency of the AA genotype was increased in adult APS ( P = 0.014). APS2/3 patients with AITD and the TNF-α -308 AA genotype showed the highest prevalence of thyroid autoantibodies. HLA-DRB1*03 and TNF-α -308*A alleles were strongly associated in patients with APS 2/3 (87.5%, P < 0.00001). In contrast, the AIRE R257X and 13bpdel mutations were not observed in patients with adult APS.
Finally, a deficiency in the DNase enzyme, and thereby a failure to remove DNA from nuclear antigens, promotes disease susceptibility to autoimmune disorders. Recent studies examined in patients with AITD and adult APS whether a reduced DNase activity is associated with sequence variations in the DNASE1 gene. In patients with adult APS, a novel mutation (1218G>A, exon 5) and multiple polymorphisms were identified in the DNASE1 gene. The allele frequency of the mutation was increased in patients vs. controls ( P < 0.001). In contrast to controls, the novel mutation was present in all five members of a family with adult APS and AITD, showing decreased DNase activity. The mutation resulted in the replacement of highly conserved valine with methionine at amino acid position 89 of the DNase enzyme. It was related to lowered heat stability and lowered activity of the enzyme. Mean expression of the DNASE1 mRNA in patients was 0.52 ± 0.22 and in controls 0.95 ± 0.22. The expression level of the DNASE1 gene was strongly decreased in patients, amounting to only 54.7% of that in controls ( P < 0.001). The identified new mutation and numerous polymorphisms, noted for the first time in patients with APS and AITD, may alter transcription and translation of the DNASE1 gene, thereby decreasing the stability and activity of the corresponding enzyme.
Pathophysiology of Mutation
APS1
The AIRE gene is expressed in tissues that are involved in the maturation of the immune system such as thymus, lymph nodes, and fetal liver. It is expressed in epithelial antigen-presenting cells in the thymus where it is involved in the central induction of self-tolerance. Thus, the AIRE gene is an important mediator of central tolerance. AIRE may regulate negative selection of organ-specific T cells. The AIRE gene encodes a 545 amino acid protein of 57.5 kDa, which comprises several domains involved in nuclear transport, DNA binding, homomultimerization, and transcriptional activity. AIRE shows several motives indicative of a transcription factor, and it includes two zinc fingers. AIRE upregulates transcription of certain organ-specific self-antigens in medullary thymic epithelial cells and plays a role in the negative selection of organ-specific thymocytes. At least three splice variant mRNAs products have been described, including one resulting in a premature stop codon in the AIRE protein and a transcript predicted to be a candidate for nuclear-mediated decay (NMD). The mutated AIRE gene results in defect AIRE proteins, causing autoimmune destruction of target organs by disturbing the immunological tolerance of the patients. Many AIRE mutations alter the nucleus-cytoplasm distribution of AIRE , thereby disturbing its association with nuclear dots and cytoplasmic filaments. The R257X mutation results in a stop codon instead of CGA (coding for arginine), leading to a truncated regulator protein. Because APS1 patients homozygous for R257X display a considerable phenotypic disease variation, further genetic or environmental factors may determine the manifestation of the syndrome.
APS2/3
The gene products of the HLA class II genes are involved in immune reactions. The different HLA class II alleles are characterized by different affinities for peptides. As a consequence, some autoantigenic peptides may be recognized by T lymphocyte receptors, whereas others may not. The CTLA-4 gene encodes a negative regulator of T-cell activation, which is expressed on the surface of activated T lymphocytes ( Table 25.2 ). It is involved in the interaction between T lymphocytes and antigen-presenting cells, APCs. APCs present to the T lymphocyte receptor an antigenic peptide bound to an HLA class II protein on the cell surface, thus activating T lymphocytes. Further, costimulatory signals on the APC surface interact with receptors (e.g., CTLA-4 ) on the surface of CD4 + T lymphocytes during antigen presentation. CTLA-4 downregulates T lymphocyte activation. CTLA-4 polymorphisms are associated with several autoimmune disorders, particularly with AITD but also with AD. In contrast, findings are inconsistent with respect to the association of CTLA-4 and T1D suggesting a weak effect. A 3′UTR (AT) n microsatellite polymorphism with longer and shorter repeats of AT may be related to autoimmunity while longer repeats are associated with decreased inhibitory function of CTLA-4 . Longer repeats were correlated with a shorter half-life of the CTLA-4 mRNA than shorter repeats. The CTLA-4 AT repeat affects the inhibitory function of CTLA-4 in that the long AT repeat allele is associated with a reduced control of T-cell proliferation in patients with GD.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


