results. Sometimes, the mutated proteins are not properly produced, secreted into the blood, or located in the physiologic site. When they do circulate in substantial amounts, the variant protein often circulates with impaired function. When a single base change is phenotypically silent, this is commonly referred to as a single nucleotide polymorphism (SNP). Outside the actual coding region, it is even more likely that nucleotide substitutions are silent. Notable exceptions are mutations in binding sites for transcription factors that regulate gene activity, mutations that affect splicing of the immature RNA, and mutations that may alter the stability of the RNA.
50% of patients, genetic risk factors such as heterozygosity for mutations in coagulation inhibitors or cofactors are found.16
Table 10.1 Characteristics of human coagulation factor genes | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 10.2 Genetic models of thrombosis and hemostasis | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
bleeding phenotype. Mutations associated with this phenotype include frameshift, splice site, promoter, and missense mutations, with the total number of mutations now reaching more than 110.76 The situation is less clear for mutations associated with measurable levels of factor VII activity, as many are asymptomatic.
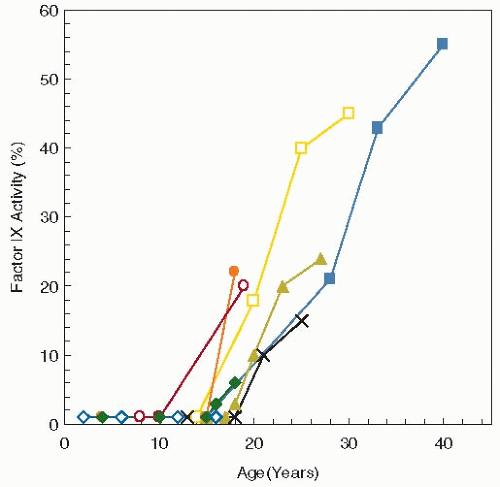 FIGURE 10.1 Factor IX levels measured over time in eight individuals with Leyden variant hemophilia B. These mutations in the promoter result in severe factor IX deficiency until puberty when factor IX levels rise gradually to approximately 30%. (Data from Briet E, Bertina RM, van Tilburg NH, et al. Hemophilia B Leyden: a sexlinked hereditary disorder that improves after puberty. (N Engl J Med 1982;306:788-790.) |
sequence and two partial deletions of the factor VIII gene were described.140 The finding of four distinctly different mutations in the factor VIII gene of otherwise similar patients was the first hint that a large variety of mutations exist in the factor VIII gene in patients with hemophilia.
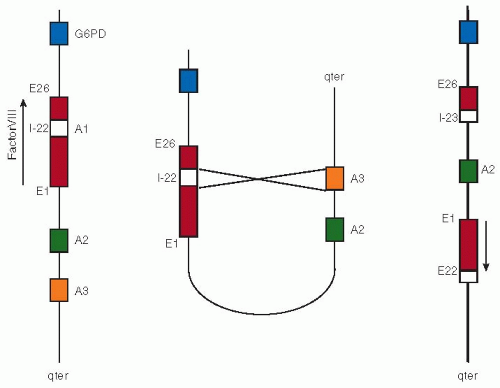 FIGURE 10.2 Factor VIII intron 22 inversion. The left panel depicts the normal alignment of the factor VIII gene on the X chromosome. Intron 22 (white box) of the factor VIII gene harbors a copy of the so-called A1 gene, whereas highly homologous A2 and A3 genes are located toward the tip of the X chromosome (indicated as qter). The middle panel shows alignment of the A1 gene with the A3 gene as it may occur during male spermatogenesis. When this alignment of A1 with A2 (or alternatively A3) results in a nonhomologous recombination, a rearrangement results as depicted at the right panel. In this rearranged X chromosome, the integrity of the factor VIII gene is completely disrupted because it is cut in two parts that are now aligned in opposite directions. |
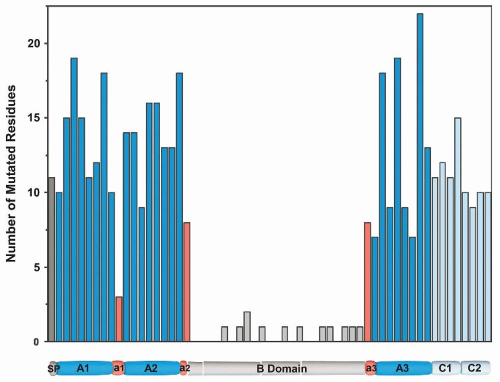 FIGURE 10.3 Graphic representation of missense mutation within factor VIII. Mutations in factor VIII leading to hemophilia A are presented in 40 residue blocks spanning the molecule. Mutations were derived from the hemophilia A mutation database (http://hadb.org.uk) with nonsense mutations excluded. (J. Coagul Disord 2010;2:19-27, with permission.) |
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


